
Structural Basis of 2-Phenylamino-4-phenoxyquinoline Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors

 , ,
, ,
Abstract
:
1. Introduction
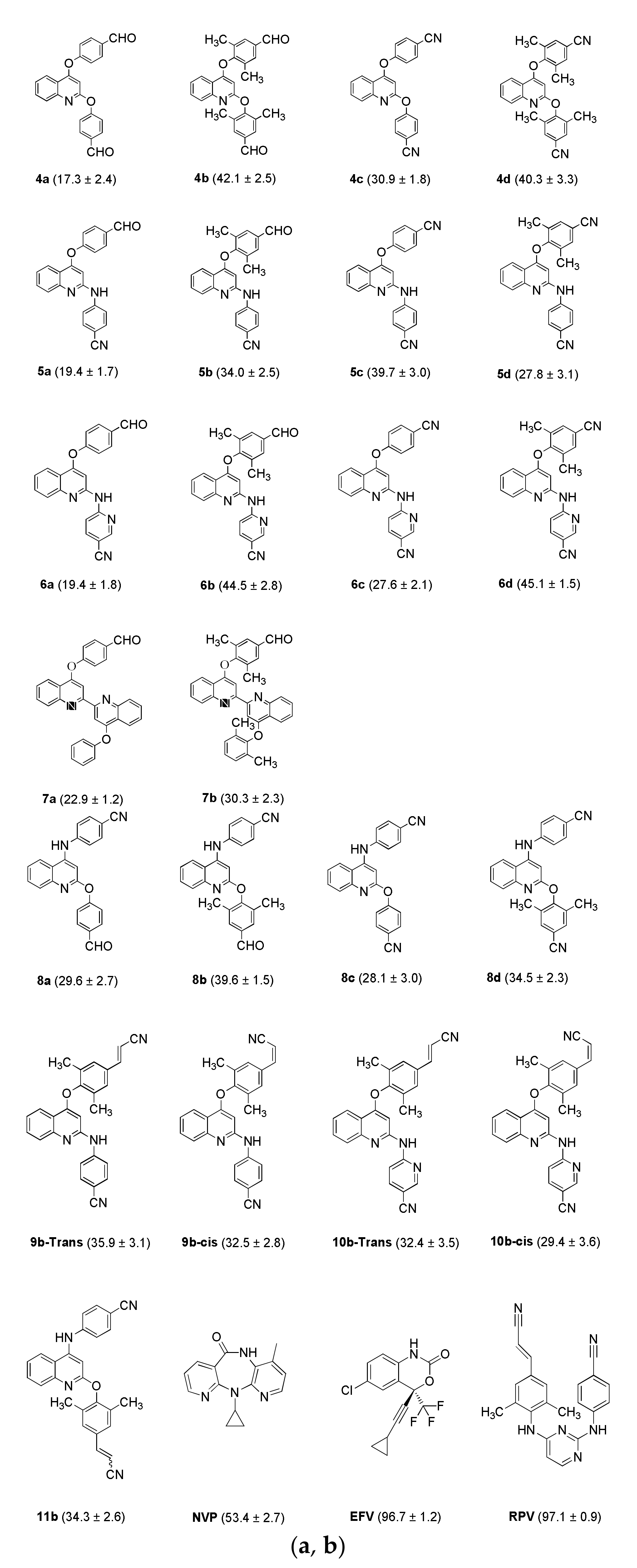
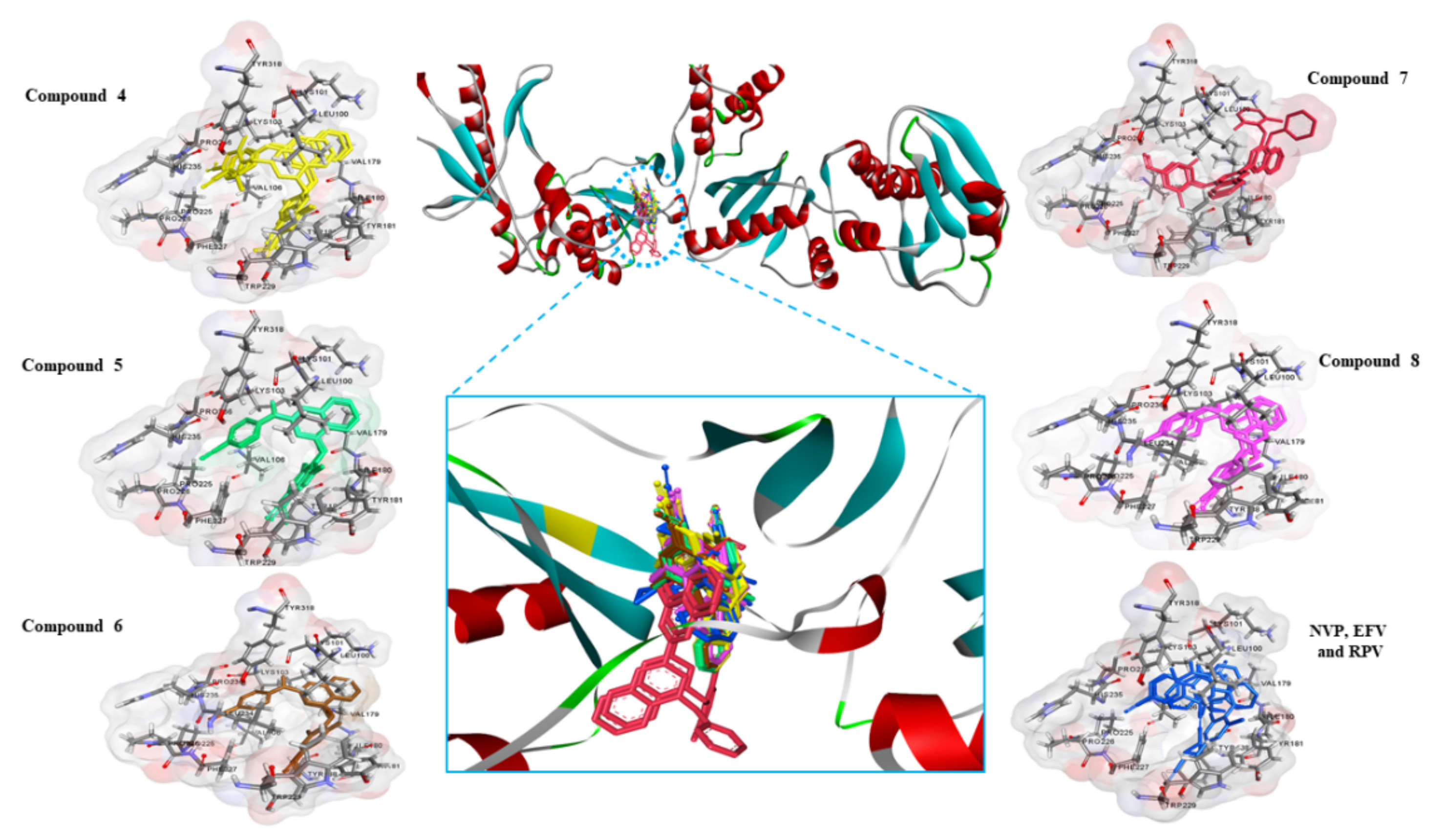
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Molecular Docking Studies
3.3. Pharmacokinetic Parameter Calculation
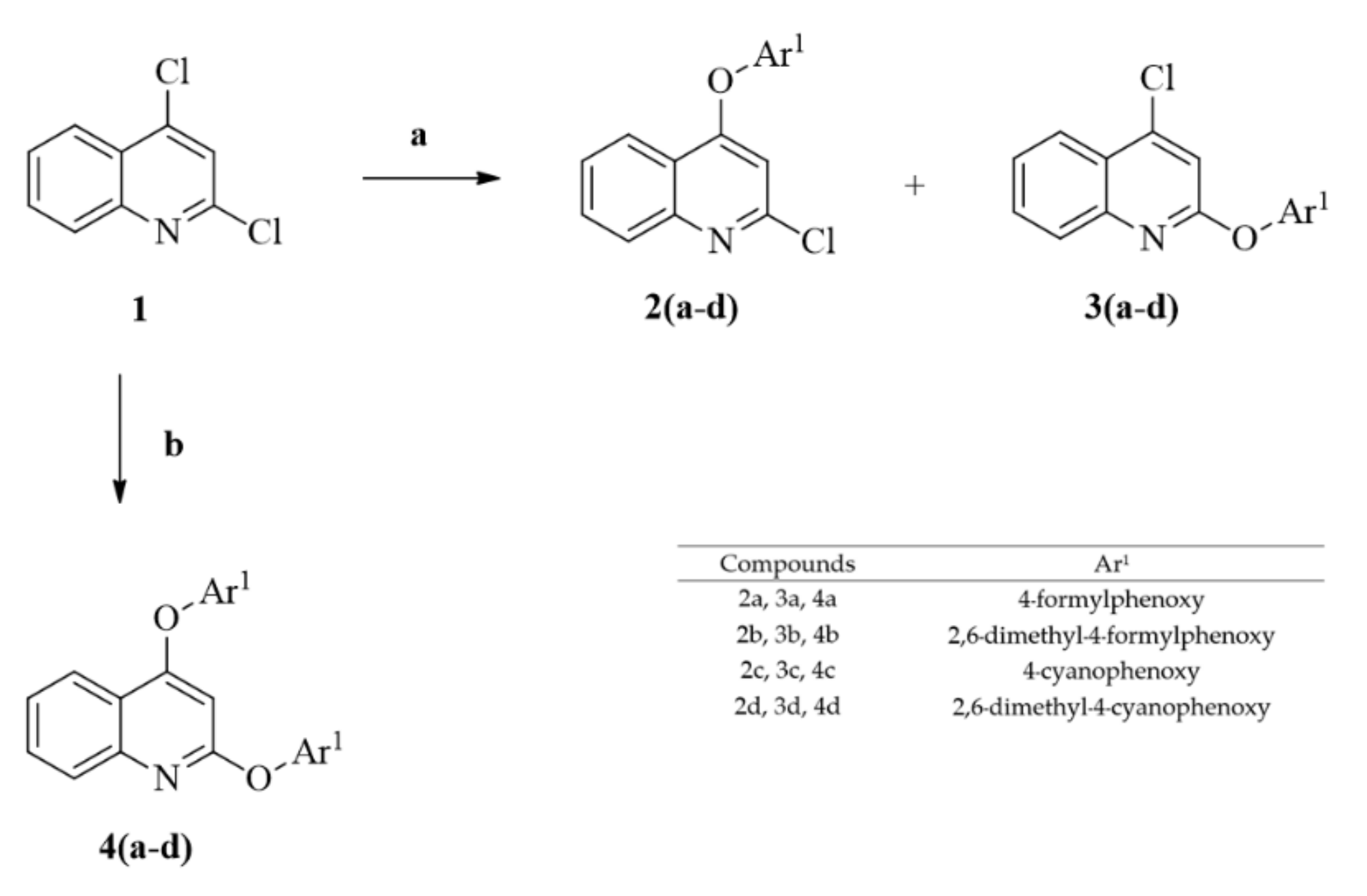
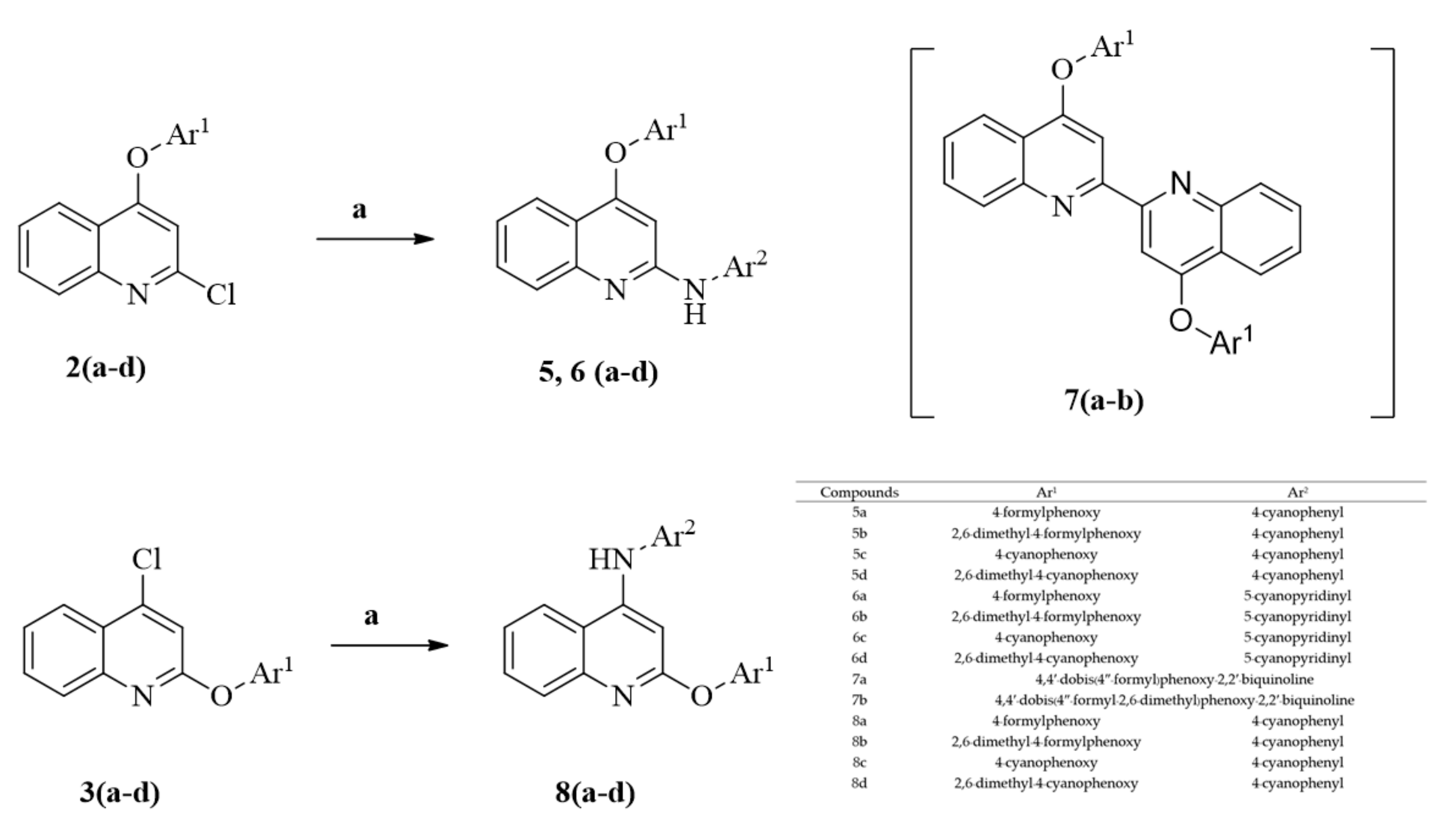
3.4. Synthesis
3.5. HIV-1 RT Inhibition Assay
3.6. Cytotoxic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Abadi, A.H.; Hegazy, G.H.; El-Zaher, A.A. Synthesis of novel 4-substituted-7-trifluoromethylquinoline derivatives with nitric oxide releasing properties and their evaluation as analgesic and anti-inflammatory agents. Bioorg. Med. Chem. 2005, 13, 5759–5765. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.N.; Thavaselvam, D.; Kaushik, M.P. Synthesis and antimalarial evaluation of novel pyridine quinoline hybrids. Med. Chem. Res. 2008, 17, 487–494. [Google Scholar] [CrossRef]
- Kumar, S.; Bawa, S.; Gupta, H. Biological Activities of Quinoline Derivatives. Mini Rev. Med. Chem. 2009, 9, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Karthikeyan, C.; Hari Narayana Moorthy, N.S.; Deora, G.S.; Solomon, V.R.; Lee, H.; Trivedi, P. Design, synthesis and biological evaluation of some novel 3-cinnamoyl-4-hydroxy-2H-chromen-2-ones as antimalarial agents. Med. Chem. Res. 2012, 21, 1780–1784. [Google Scholar] [CrossRef]
- Assefa, H.; Kamath, S.; Buolamwini, J.K. 3D-QSAR and docking studies on 4-anilinoquinazoline and 4-anilinoquinoline epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. J. Comput. Aided Mol. Des. 2003, 17, 475–493. [Google Scholar] [CrossRef]
- Mathes, T.; Pieper, D.; Antoine, S.-L.; Eikermann, M. Adherence-enhancing interventions for highly active antiretroviral therapy in HIV-infected patients—A systematic review. HIV Med. 2013, 14, 583–595. [Google Scholar] [CrossRef]
- Slama, L.; Li, X.; Brown, T.; Jacobson, L.P.; Pialoux, G.; Macatangay, B.; Bolan, R.K.; Phair, J.; Palella, F.J., Jr. Increases in duration of first highly active antiretroviral therapy over time (1996–2009) and associated factors in the multicenter AIDS cohort study. JAIDS J. Acquir. Immune Defic. Syndr. 2014, 65, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Asahchop, E.L.; Wainberg, M.A.; Sloan, R.D.; Tremblay, C.L. Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5000–5008. [Google Scholar] [CrossRef] [Green Version]
- Domaoal, R.A.; Demeter, L.M. Structural and biochemical effects of human immunodeficiency virus mutants resistant to non-nucleoside reverse transcriptase inhibitors. Int. J. Biochem. Cell Biol. 2004, 36, 1735–1751. [Google Scholar] [CrossRef]
- Mehellou, Y.; Clercq, E.D. Twenty-Six Years of anti-hiv drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef]
- Delaugerre, C.; Rohban, R.; Simon, A.; Mouroux, M.; Tricot, C.; Agher, R.; Huraux, J.M.; Katlama, C.; Calvez, V. Resistance profile and cross-resistance of HIV-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J. Med. Virol. 2001, 65, 445–448. [Google Scholar] [CrossRef]
- Ludovici, D.W.; De Corte, B.L.; Kukla, M.J.; Ye, H.; Ho, C.Y.; Lichtenstein, M.A.; Kavash, R.W.; Andries, K.; Béthune, M.-P.; Azijn, H.; et al. Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogues. Bioorg. Med. Chem. Lett. 2001, 11, 2235–2239. [Google Scholar] [CrossRef]
- Janssen, P.A.; Lewi, P.J.; Arnold, E.; Daeyaert, F.; de Jonge, M.; Heeres, J.; Koymans, L.; Vinkers, M.; Guillemont, J.; Pasquier, E.; et al. In Search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)- 2-Cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino] benzonitrile (R278474, Rilpivirine). J. Med. Chem. 2005, 48, 1901–1909. [Google Scholar] [CrossRef]
- Makarasen, A.; Kuno, M.; Patnin, S.; Reukngam, N.; Khlaychan, P.; Deeyohe, S.; Intachote, P.; Saimanee, B.; Sengsai, S.; Boonsri, P.; et al. Molecular Docking Studies and Synthesis of Amino-oxydiarylquinoline Derivatives as Potent Non-nucleoside HIV-1 Reverse Transcriptase Inhibitors. Drug. Res. 2019, 69, 671–682. [Google Scholar]
- Patnin, S.; Makarasen, A.; Kuno, M.; Deeyohe, S.; Techasakul, S.; Chaivisuthangkura, A. Binding interaction of potent HIV-1 NNRTIs, amino-oxy-diarylquinoline with the transport protein using spectroscopic and molecular docking. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2020, 233, 118159–118165. [Google Scholar] [CrossRef]
- Kretsos, K.; Miller, M.A.; Zamora-Estrada, G.; Kasting, G.B. Partitioning, diffusivity and clearance of skin permeants in mammalian dermis. Int. J. Pharm. 2008, 346, 64–79. [Google Scholar] [CrossRef]
- Hartmann, T.; Schmitt, J. Lipophilicity–beyond octanol/water: A short comparison of modern technologies. Drug Discov. Today Technol. 2004, 4, 431–439. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar]
- Palm, K.; Luthman, K.; Unge, A.-L.; Strandlund, G.; Artursson, P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci. 1996, 85, 32–39. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Waterbeemd, H.V.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding characteristics. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef]
- Ren, J.; Nichols, C.E.; Stamp, A.; Chamberlain, P.P.; Ferris, R.; Weaver, K.L.; Short, S.A.; Stammers, D.K. Structure insights into mechanisms of non-nucleoside drug resistance for HIV-1 reverse transcriptases mutated at codons 101 or 138. FEBS J. 2006, 273, 3850–3860. [Google Scholar] [CrossRef]
- Díaz-Delfín, J.; Domingo, P.; Mateo, M.G.; Gutierrez, M.M.; Domingo, J.C.; Giralt, M.; Villarroya, F. Effects of Rilpivirine on Human Adipocyte Differentiation, Gene Expression, and Release of Adipokines and Cytokines. Antimicrob. Agents Chemother. 2012, 56, 3369–3375. [Google Scholar] [CrossRef] [Green Version]
- Hecht, M.; Harrer, T.; Büttner, M.; Schwegler, M.; Erber, S.; Fietkau, R.; Distel, L.V. Cytotoxic effect of efavirenz is selective against cancer cells and associated with the cannabinoid system. AIDS 2013, 27, 2031–2040. [Google Scholar] [CrossRef]
- Mangiacasale, R.; Pittoggi, C.; Sciamanna, I.; Careddu, A.; Mattei, E.; Lorenzini, R.; Travaglini, L.; Landriscina, M.; Barone, C.; Nervi, C.; et al. Exposure of normal and transformed cells to nevirapine, a reverse transcriptase inhibitor, reduces cell growth and promotes differentiation. Oncogene 2003, 22, 2750–2761. [Google Scholar] [CrossRef] [Green Version]
- Sinibaldi-Vallebona, P.; Lavia, P.; Garaci, E.; Spadafora, C. A role for endogenous reverse transcriptase in tumorigenesis and as a target in differentiating cancer therapy. Genes Chromosomes Cancer 2006, 45, 1–10. [Google Scholar] [CrossRef]
- Kuroda, D.G.; Bauman, J.D.; Challa, J.R.; Patel, D.; Troxler, T.; Das, K.; Arnold, E.; Hochstrasser, R.M. Snapshot of the equilibrium dynamics of a drug bound to HIV-1 reverse transcriptase. Nat. Chem. 2013, 5, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDockTools4 and AutoDock4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42729. [Google Scholar] [CrossRef] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ukkonen, P.; Korpela, J.; Suni, J.; Hedman, K. Inactivation of human immunodeficiency virus in serum specimens as a safety measure for diagnostic immunoassays. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 518–523. [Google Scholar] [CrossRef]
- Suzuki, K.; Craddock, B.P.; Okamoto, N.; Kano, T.; Steigbigel, R. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J. Virol. Methods 1993, 44, 189–198. [Google Scholar] [CrossRef]
- Doyly, A.; Griffiths, J.B. Mammalian Cell Culture-Essential Techniques; Wiley & Sons: Chichester, UK, 1997. [Google Scholar]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Binding Energy (kcal/mol) a | NOC b | Interaction Amino Acids 3 Å (4G1Q) | |
|---|---|---|---|---|
| H-Bond | Pi–Pi Stacking | |||
| 4a | −11.5 ± 0 | 83 ± 5 | HIS235 | - |
| 4b | −12.6 ± 0 | 21 ± 1 | - | TYR188 TRP229 |
| 4c | −12.3 ± 0 | 137 ± 3 | LYS101 | TRP229 TYR318 |
| 4d | −13.2 ± 0 | 62 ± 6 | TYR188 TRP229 | TYR188 TRP229 |
| 5a | −12.1 ± 0 | 64 ± 7 | LYS101 HIS235 | TYR318 |
| 5b | −13.2 ± 0 | 81 ± 9 | LYS101 HIS235 | TYR181 TYR318 |
| 5c | −12.5 ± 0 | 87 ± 7 | LYS101 HIS235 | TRP229 TYR318 |
| 5d | −13.7 ± 0 | 96 ± 5 | LYS101 HIS235 | TYR318 |
| 6a | −13.7 ± 0 | 68 ± 6 | LYS101 HIS235 | TYR188 TYR318 |
| 6b | −12.7 ± 0 | 70 ± 75 | LYS101 HIS235 PRO236 | TYR188 TRP229 TYR318 |
| 6c | −12.0 ± 0 | 61 ± 4 | LYS101 HIS235 | TYR188 TYR318 |
| 6d | −13.1 ± 0 | 74 ± 1 | LYS101 HIS235 PRO236 | TYR188 TRP229 TYR318 |
| 7a | −11.7 ± 0 | 59 ± 3 | TYR318 | TYR181 |
| 7b | −12.1 ± 0 | 12 ± 2 | TYR318 | TYR181 |
| 8a | −12.0 ± 0 | 76 ± 5 | HIS235 | TYR181 TRP229 TYR318 |
| 8b | −12.7 ± 0 | 76 ± 10 | LYS101 HIS235 PRO236 | TYR181 TYR188 TRP229 TYR318 |
| 8c | −12.3 ± 0 | 125 ± 6 | LYS101 HIS235 | TYR181 TRP229 TYR318 |
| 8d | −13.2 ± 0 | 84 ± 6 | LYS101 HIS235 PRO236 | TYR181 TYR188 TRP229 TYR318 |
| 9b-trans | −14.8 ± 0 | 110 ± 6 | LYS101 | TRP229 |
| 9b-cis | −14.0 ± 0 | 47 ± 2 | LYS101 HIS235 | TYR318 |
| 10b-trans | −14.3 ± 0 | 90 ± 5 | LYS101 HIS235 PRO236 | TYR181 TRP229 TYR318 |
| 10b-cis | −13.5 ± 0 | 45 ± 6 | LYS101 HIS235 | TYR181 TYR318 |
| 11b | −14.2 ± 0 (T) | 95 ± 7 | LYS101 HIS235 | TYR181 TYR188 TRP229 |
| −13.8 ± 0 (C) | 45 ± 4 | LYS101 PRO236 | TYR181 TRP229 | |
| NVP | −7.9 ± 0 | 122 ± 0 | LYS101 | - |
| EVF | −9.2 ± 0 | 132 ± 4 | LYS101 | - |
| RVP | −12.5 ± 0 | 129 ± 9 | LYS101 | TYR181 TRP229 |
| Ligands | Number of H-Bond Acceptors | Number of H-Bond Donors | LogP | Number of Rotatable Bonds | Molecular Weight (g/mol) | TPSA (Å2) |
|---|---|---|---|---|---|---|
| 4a | 5 | 0 | 2.75 | 6 | 369 | 65.49 |
| 4b | 5 | 0 | 3.58 | 6 | 425 | 65.49 |
| 4c | 5 | 0 | 2.75 | 4 | 363 | 78.93 |
| 4d | 5 | 0 | 3.58 | 4 | 419 | 78.93 |
| 5a | 5 | 1 | 2.75 | 4 | 365 | 75.01 |
| 5b | 5 | 1 | 3.17 | 4 | 393 | 75.01 |
| 5c | 4 | 1 | 2.75 | 4 | 362 | 81.73 |
| 5d | 4 | 1 | 3.17 | 4 | 390 | 81.73 |
| 6a | 5 | 1 | 1.73 | 5 | 366 | 87.90 |
| 6b | 5 | 1 | 2.15 | 5 | 394 | 87.90 |
| 6c | 5 | 1 | 1.73 | 4 | 363 | 94.62 |
| 6d | 5 | 1 | 2.15 | 4 | 391 | 94.62 |
| 7a | 5 | 0 | 3.92 | 6 | 468 | 61.31 |
| 7b | 5 | 0 | 4.65 | 6 | 524 | 61.31 |
| 8a | 4 | 1 | 2.75 | 5 | 365 | 75.01 |
| 8b | 4 | 1 | 3.17 | 5 | 393 | 75.01 |
| 8c | 4 | 1 | 2.75 | 4 | 362 | 81.73 |
| 8d | 4 | 1 | 3.17 | 4 | 390 | 81.73 |
| 9 b-trans | 4 | 1 | 3.51 | 5 | 416 | 81.73 |
| 9 b-cis | 4 | 1 | 3.51 | 5 | 416 | 81.73 |
| 10 b-trans | 5 | 1 | 2.50 | 5 | 417 | 94.62 |
| 10 b-cis | 5 | 1 | 2.50 | 5 | 417 | 94.62 |
| 11 b-trans | 4 | 1 | 3.51 | 4 | 416 | 81.73 |
| 11 b-cis | 4 | 1 | 3.51 | 4 | 416 | 81.73 |
| NVP | 1 | 1 | 2.14 | 1 | 266 | 63.57 |
| EVF | 1 | 1 | 3.61 | 1 | 315 | 38.33 |
| RPV | 5 | 3 | 2.37 | 5 | 366 | 97.42 |
| Compounds | IC50 (µM) b |
|---|---|
| 6b | 1.93 |
| 6d | 1.22 |
| NVP | 1.05 |
| EFV | 0.06 |
| RPV | 0.06 |
| Compounds (Molecular Weight) | Cell Lines a (IC50 [µM]) b | ||||
|---|---|---|---|---|---|
| HepG2 a | MOLT-3 a | HuCCA-1 a | A549 a | MRC-5 a | |
| 4a (369.4) | 90.9 ± 2.1 | 25.1 ± 2.6 | 48.1 ± 1.7 | %C = 40.0 c | 84.3 ± 9.6 |
| 4b (425.5) | 50.6 ± 1.2 | 61.6 ± 0.7 | 11.1 ± 0.7 | 38.5 ± 1.3 | 60.4 ± 2.0 |
| 4c (363.4) | %C = 41.4 c | %C = 11.0 c | %C = 15.0 c | %C = 12.0 c | 107.6 ± 6.4 |
| 4d (419.5) | 92.2 ± 6.8 | %C = 13.0 c | 83.5 ± 11.1 | 55.5 ± 5.7 | %C = 26.9 c |
| 5a (365.4) | 73.2 ± 7.7 | 15.6 ± 3.6 | 29.7 ± 2.0 | 121.7 ± 4.5 | 107.9 ± 4.7 |
| 5b (393.4) | 65.9 ± 6.1 | 11.8 ± 1.6 | 29.4 ± 2.0 | 51.2 ± 3.0 | 69.8 ± 4.3 |
| 5c (362.4) | %C = 44.8 c | %C = 6.0 c | %C = 23.0 c | %C = 26.0 c | %C = 12.0 c |
| 5d (390.4) | %C = 38.2 c | 48.8 ± 41.0 | %C = 3.0 c | %C = 21.0 c | %C = 7.4 c |
| 6a (366.4) | 23.7 ± 1.0 | 5.13 ± 1.5 | 14.9 ± 1.1 | %C = 42.0 c | 18.3 ± 3.9 |
| 6b (393.4) | 92.8 ± 11.8 | 12.7 ± 1.1 | 125.8 ± 1.8 | 107.9 ± 1.2 | %C = 26.1 c |
| 6c (363.4) | 101.7 ± 0.7 | 14.1 ± 0.4 | 22.9 ± 0.9 | 14.0 ± 2.2 | 69.2 ± 12.4 |
| 6d (391.4) | %C = 0.0 c | %C = 27.0 c | %C = 0.0 c | %C = 8.0 c | %C = 0.0 c |
| 7a (496.5) | 69.8 ± 11.8 | 74.2 ± 6.3 | 88.2 ± 4.4 | 69.5 ± 3.3 | %C = 33.9 c |
| 7b (552.6) | %C = 0.1 c | %C = 6.0 c | %C = 4.0 c | %C = 15.0 c | %C = 4.2 c |
| 8a (365.4) | 85.3 ± 3.5 | 21.5 ± 3.3 | 22.7 ± 1.9 | 111.2 ± 5.6 | 119.0 ± 5.7 |
| 8b (393.4) | 105.8 ± 9.7 | 22.3 ± 1.9 | %C = 43.0 c | %C = 37.0 c | %C = 9.7 c |
| 8c (393.4) | %C = 40.3 c | %C = 2.1 c | %C = 25.8 c | %C = 29.0 c | %C = 17.2 c |
| 8d (393.4) | %C = 32.0 c | 67.4 ± 3.1 | %C = 18.4 c | %C = 29.1 c | %C = 15.5 c |
| 9b-trans (416.5) | %C = 42.9 c | %C = 19.0 c | %C = 8.0 c | %C = 8.0 c | %C = 28.4 c |
| 9b-cis (416.5) | %C = 9.8 c | 52.8 ± 2.1 | %C = 10.0 | %C = 1.0 c | %C = 0.0 c |
| 10b-trans (417.5) | %C = 1.0 c | %C = 2.0 c | %C = 0.0 c | %C = 1.0 c | %C = 0.0 c |
| 10b-cis (417.5) | %C = 0.0 c | %C = 11.0 c | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c |
| 11b (416.5) | %C = 3.7 c | %C = 19.0 c | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c |
| NVP (266.3) | %C = 15.4 c | %C = 29.0 c | %C = 11.0 c | %C = 18.0 c | %C = 1.0 c |
| EFV (315.7) | 86.6 ± 3.9 | 24.6 ± 1.2 | 59.6 ± 2.4 | 60.2 ± 1.8 | 95.6 ± 6.0 |
| RPV (366.4) | 87.4 ± 0.1 | 4.3 ± 0.5 | 61.3 ± 1.7 | %C = 42.0 c | %C = 14.4 c |
| Doxorubicin (543.5) | 0.6 ± 0.0 | 0.02 ± 0.00 | 0.9 ± 0.0 | 0.6 ± 0.0 | 3.1 ± 0.5 |
| Etoposide (588.6) | 49.9 ± 1.1 | 0.03 ± 0.0 | - | - | - |
| Compounds (Molecular Weight) | Cell Lines a (IC50 [µM]) b | |||||
|---|---|---|---|---|---|---|
| MDA-MB-231 a | S102 a | HeLA a | T47-D a | H69AR a | HL-60 a | |
| 4a (369.4) | %C = 43.3 c | %C = 0.4 c | 103.5 ± 7.9 | %C = 34.0 c | %C = 0.0 c | 92.4 ± 3.1 |
| 4b (425.5) | 15.3 ± 1.6 | 89.4 ± 0.1 | 5.3 ± 0.2 | 22.5 ± 3.3 | 30.9 ± 5.7 | 5.7 ± 2.0 |
| 4c (363.4) | 92.5 ± 4.2 | %C = 23.7 c | %C = 23.0 c | %C = 27.0 c | %C = 21.0 c | %C = 15.0 c |
| 4d (419.5) | 92.5 ± 5.0 | %C = 31.5 c | %C = 40.0 c | 55.2 ± 1.5 | %C = 38.0 c | %C = 23.0 c |
| 5a (365.4) | 77.7 ± 5.9 | %C = 20.9 c | 12.6 ± 1.3 | 43.0 ± 5.3 | %C = 38.0 c | 16.6 ± 4.7 |
| 5b (393.4) | 15.9 ± 0.4 | 74.0 ± 5.3 | 3.9 ± 0.6 | 22.9 ± 0.9 | 27.6 ± 3.4 | 4.5 ± 0.2 |
| 5c (362.4) | %C = 38.4 c | %C = 0.0 c | %C = 25.0 c | %C = 45.0 c | %C = 26.0 c | %C = 2.0 c |
| 5d (390.4) | %C = 14.9 c | %C = 0.0 c | 83.4 ± 5.7 | %C = 24.0 c | %C = 0.0 c | 33.4 ± 1.7 |
| 6a (366.4) | 68.0 ± 2.0 | %C = 31.7 c | 15.4 ± 0.8 | 46.3 ± 0.2 | 64.2 ± 2.5 | 15.0 ± 1.8 |
| 6b (393.4) | 102.7 ± 1.9 | 111.5 ± 1.8 | 25.7 ± 0.8 | %C = 38.0 c | %C = 21.0 c | 20.5 ± 2.1 |
| 6c (363.4) | 66.6 ± 3.0 | %C = 44.8 c | 67.7 ± 0.6 | 47.1 ± 3.3 | 66.2 ± 2.4 | %C = 9.0 c |
| 6d (391.4) | %C = 8.3 c | %C = 0.0 c | %C = 0.0 c | %C = 1.0 c | %C = 0.0 c | %C = 13.0 c |
| 7a (496.5) | 74.1 ± 5.3 | %C = 17.6 c | 88.0 ± 4.3 | 96.1 ± 3.3 | %C = 20.0 c | %C = 25.0 c |
| 7b (552.6) | %C = 4.6 c | %C = 0.0 c | %C = 13.0 c | %C = 36.0 c | %C = 9.0 c | %C = 15.0 c |
| 8a (365.4) | 117.7 ± 5.8 | %C = 2.9 c | 112.3 ± 3.1 | %C = 0.0 c | %C = 8.4 c | 106.9 ± 1.4 |
| 8b (393.4) | 107.0 ± 3.2 | %C = 0.0 c | 42.3 ± 2.7 | %C = 38.0 c | %C = 6.0 c | 34.9 ± 5.1 |
| 8c (393.4) | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c | %C = 35.0 c | %C = 21.0 c | %C = 1.0 c |
| 8d (393.4) | %C = 32.4 c | %C = 0.0 c | 103.8 ± 4.7 | %C = 31.0 c | %C = 0.0 c | 56.6 ± 4.7 |
| 9b-trans (416.5) | %C = 24.9 c | %C = 0.0 c | %C = 38.0 c | %C = 0.0 c | %C = 14.0 c | %C = 17.0 c |
| 9b-cis (416.5) | %C = 10.0 c | %C = 0.0 c | 97.2 ± 5.1 | %C = 5.0 c | %C = 0.0 c | %C = 42.0 c |
| 10b-trans (417.5) | %C = 17.0 c | %C = 3.1 c | %C = 10.0 c | %C = 0.0 c | %C = 0.0 c | %C = 31.0 c |
| 10b-cis (417.5) | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c | %C = 0.0 c | %C = 5.0 c |
| 11b (416.5) | %C = 14.1 c | %C = 0.0 c | %C = 2.0 c | %C = 4.0 c | %C = 3.0 c | %C = 0.0 c |
| NVP (266.3) | %C = 8.9 c | %C = 3.0 c | %C = 34.0 c | %C = 14.0 c | %C = 8.0 c | %C = 3.0 c |
| EFV (315.7) | 77.7 ± 5.6 | 93.5 ± 4.7 | 46.2 ± 1.3 | 59.2 ± 0.8 | 105.0 ± 3.4 | 33.7 ± 1.1 |
| RPV (366.4) | 25.3 ± 2.9 | 22.5 ± 1.2 | 11.3 ± 1.2 | 15.0 ± 1.3 | 57.1 ± 2.0 | 11.5 ± 0.8 |
| Doxorubicin (543.5) | 2.5 ± 0.1 | 2.2 ± 0.0 | 0.4 ± 0.0 | 1.2 ± 0.0 | 34.3 ± 0.1 | 0.1 ± 0.0 |
| Etoposide (588.6) | - | - | - | - | - | 0.8 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarasen, A.; Patnin, S.; Vijitphan, P.; Reukngam, N.; Khlaychan, P.; Kuno, M.; Intachote, P.; Saimanee, B.; Sengsai, S.; Techasakul, S. Structural Basis of 2-Phenylamino-4-phenoxyquinoline Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules 2022, 27, 461. https://doi.org/10.3390/molecules27020461
Makarasen A, Patnin S, Vijitphan P, Reukngam N, Khlaychan P, Kuno M, Intachote P, Saimanee B, Sengsai S, Techasakul S. Structural Basis of 2-Phenylamino-4-phenoxyquinoline Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules. 2022; 27(2):461. https://doi.org/10.3390/molecules27020461
Chicago/Turabian StyleMakarasen, Arthit, Suwicha Patnin, Pongsit Vijitphan, Nanthawan Reukngam, Panita Khlaychan, Mayuso Kuno, Pakamas Intachote, Busakorn Saimanee, Suchada Sengsai, and Supanna Techasakul. 2022. "Structural Basis of 2-Phenylamino-4-phenoxyquinoline Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors" Molecules 27, no. 2: 461. https://doi.org/10.3390/molecules27020461
APA StyleMakarasen, A., Patnin, S., Vijitphan, P., Reukngam, N., Khlaychan, P., Kuno, M., Intachote, P., Saimanee, B., Sengsai, S., & Techasakul, S. (2022). Structural Basis of 2-Phenylamino-4-phenoxyquinoline Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules, 27(2), 461. https://doi.org/10.3390/molecules27020461