
A New Series of Indeno[1,2-c]pyrazoles as EGFR TK Inhibitors for NSCLC Therapy

 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
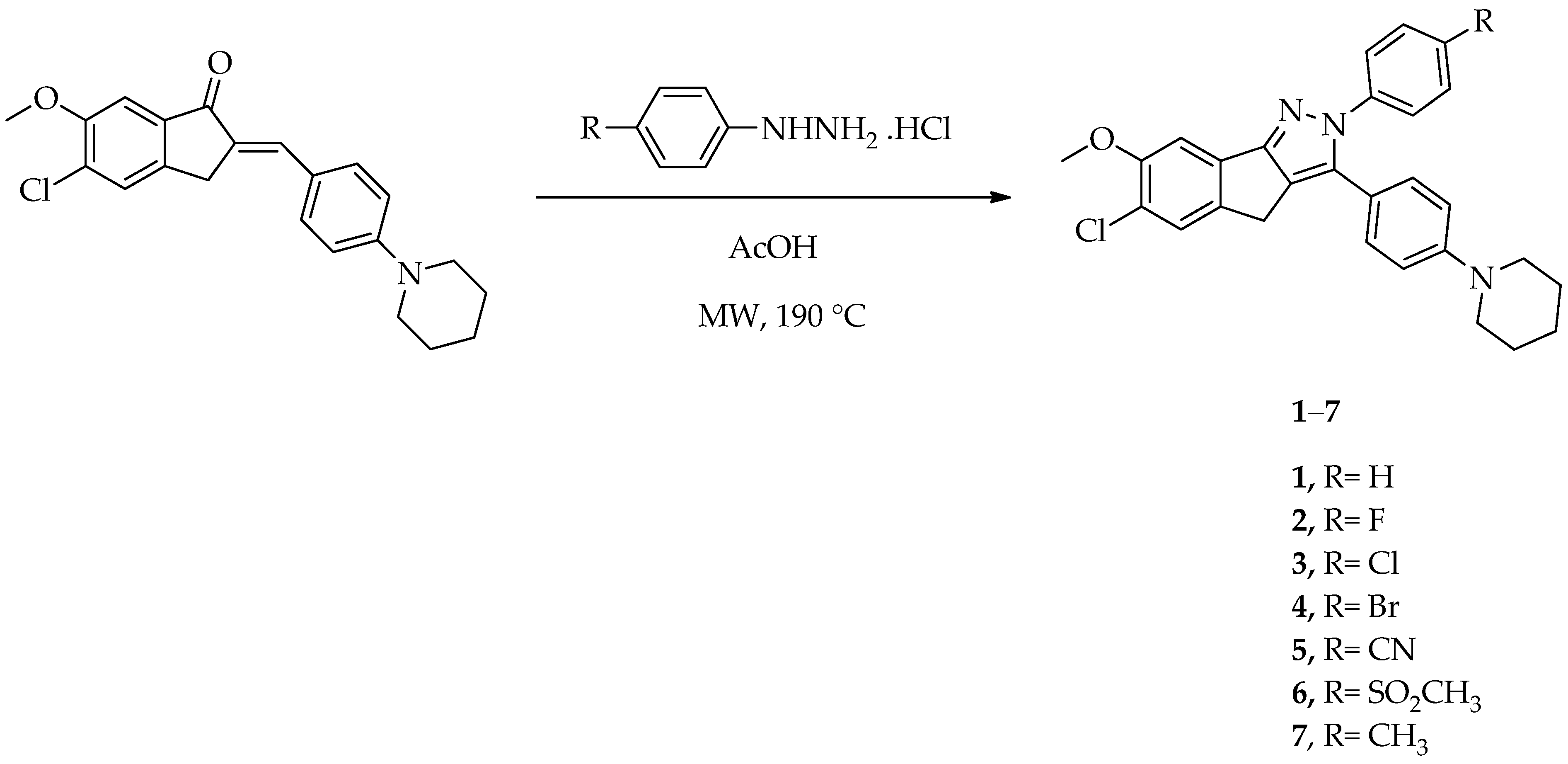
2.1. Chemistry
2.2. In Vitro Assays
2.3. In Silico Assays
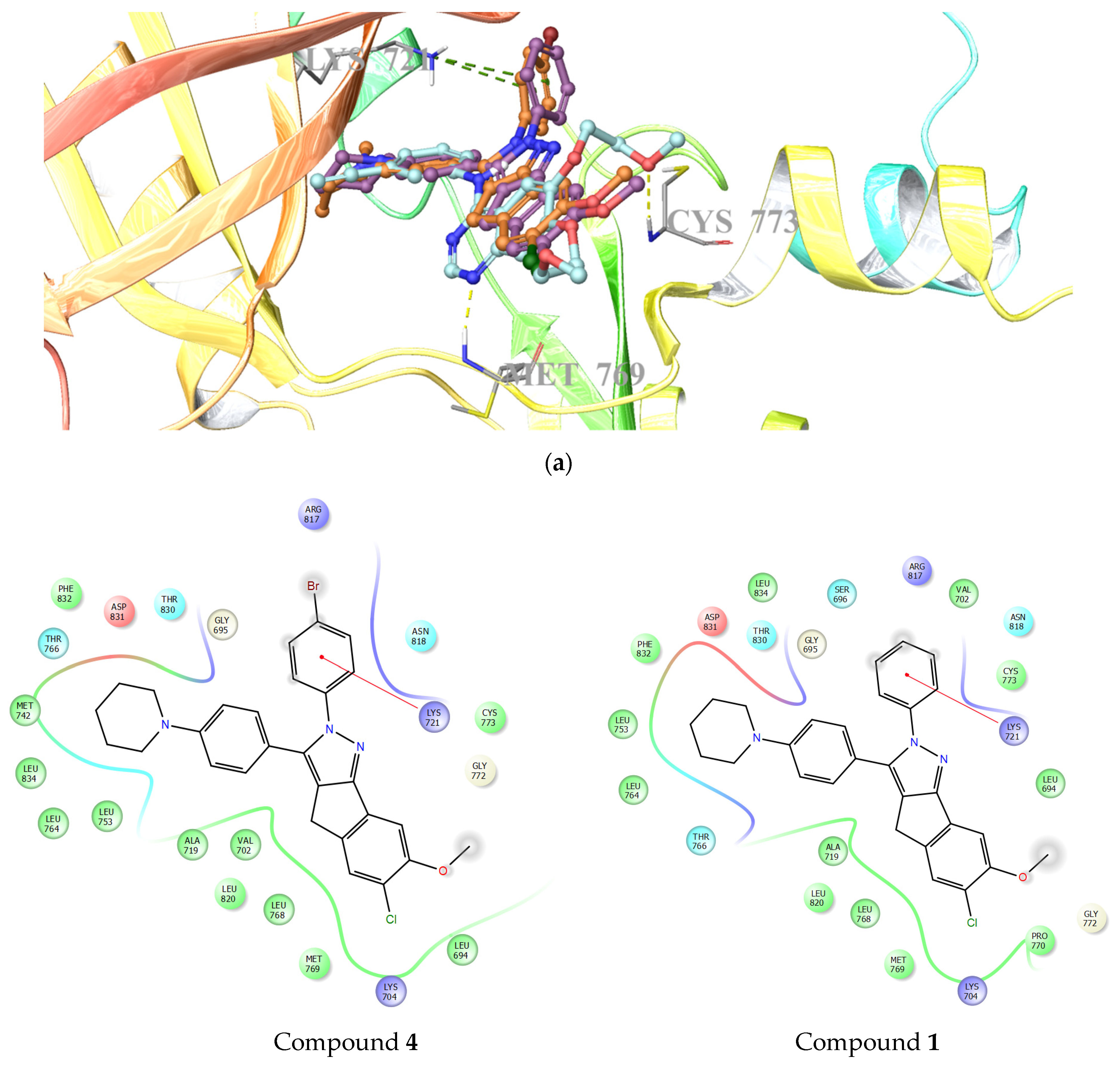
2.3.1. Molecular Docking Studies
2.3.2. In Silico Absorption, Distribution, Metabolism, and Excretion (ADME) Studies
3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of 6-Chloro-7-methoxy-2-aryl-3-[4-(piperidin-1-yl)phenyl]-2,4-dihydroindeno[1,2-c]pyrazoles (1–7)
3.2. Biochemistry
3.2.1. Cell Culture and Drug Treatments
3.2.2. Cell Viability Assay
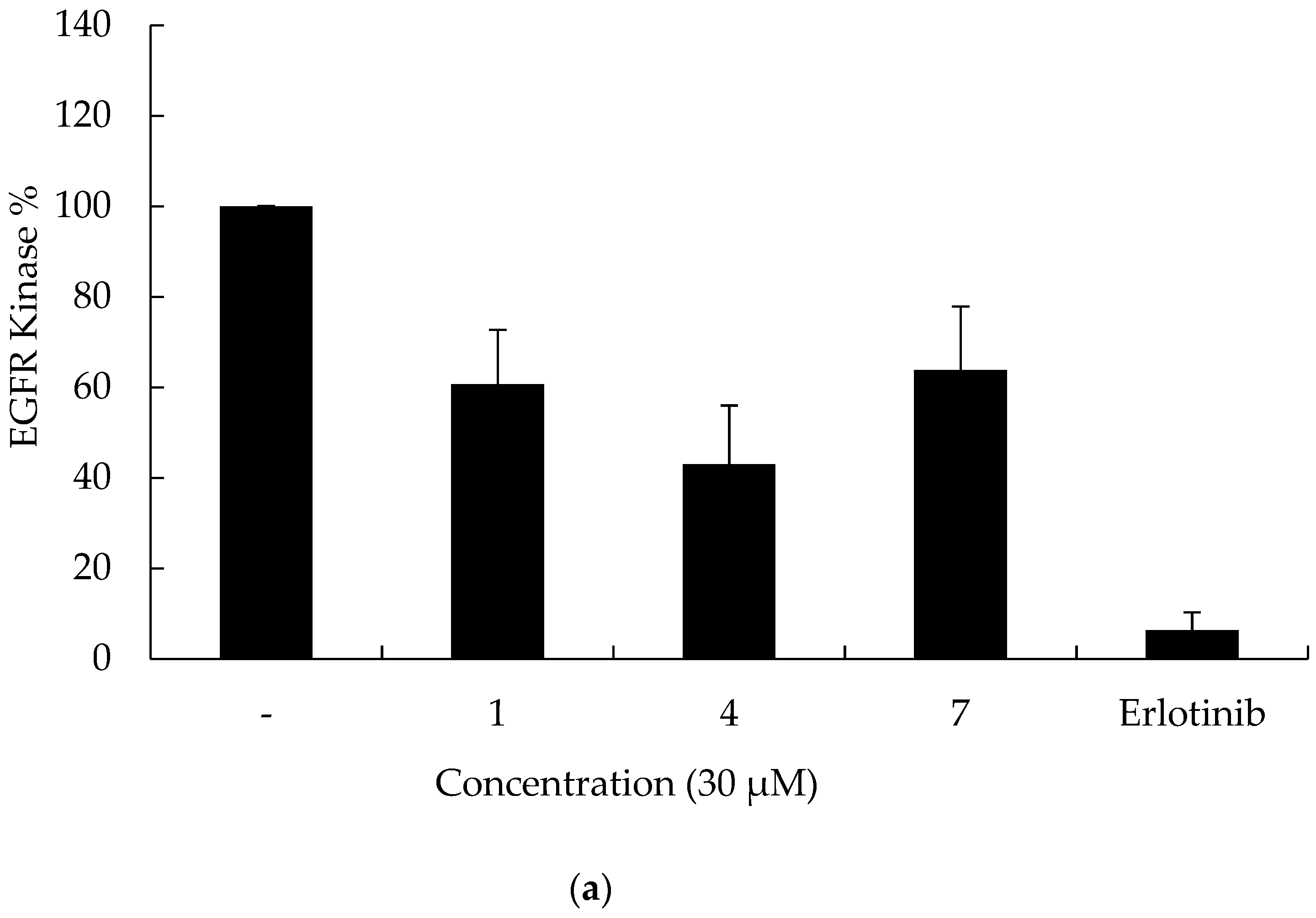
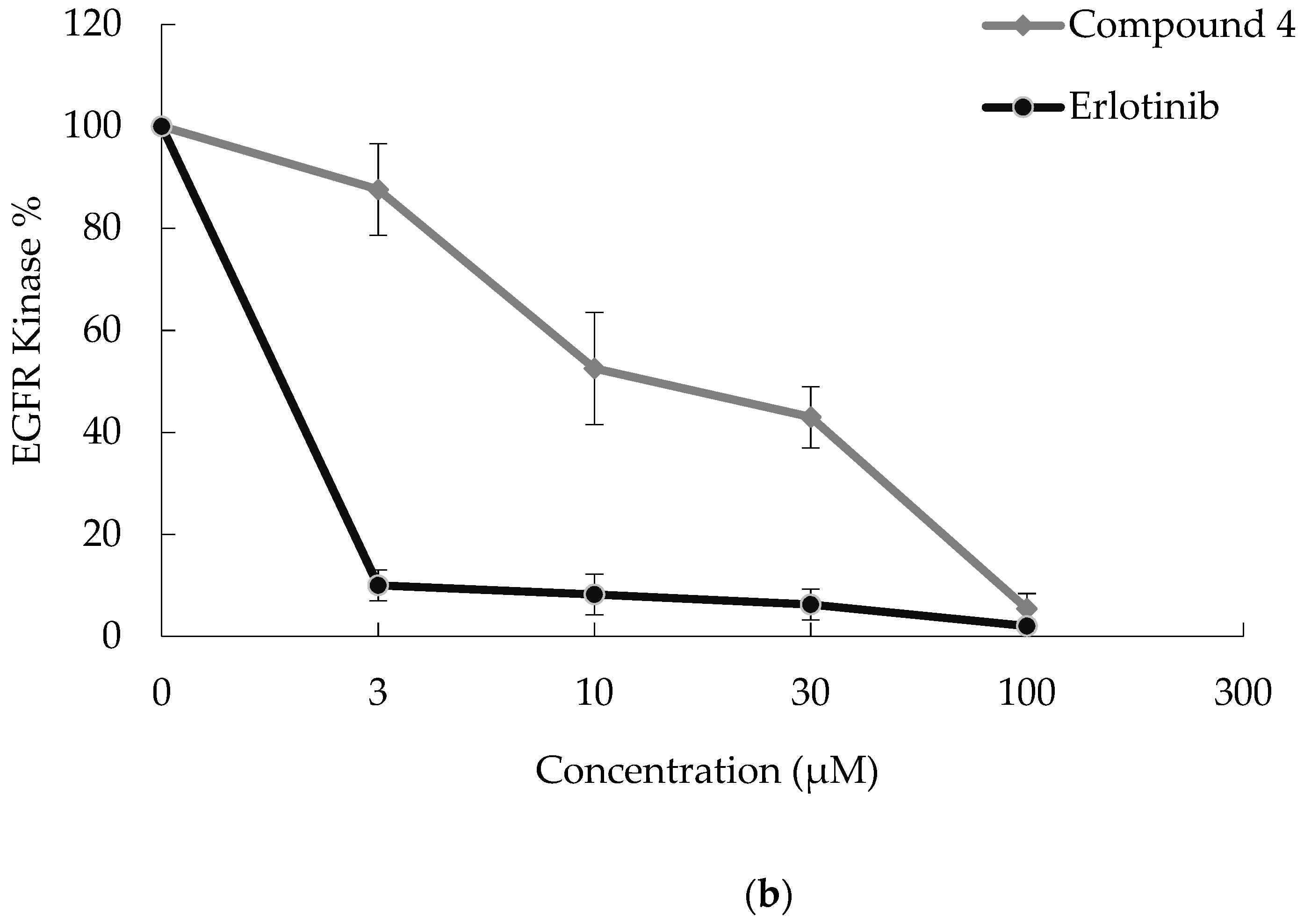
3.2.3. Kinase Inhibitory Activity
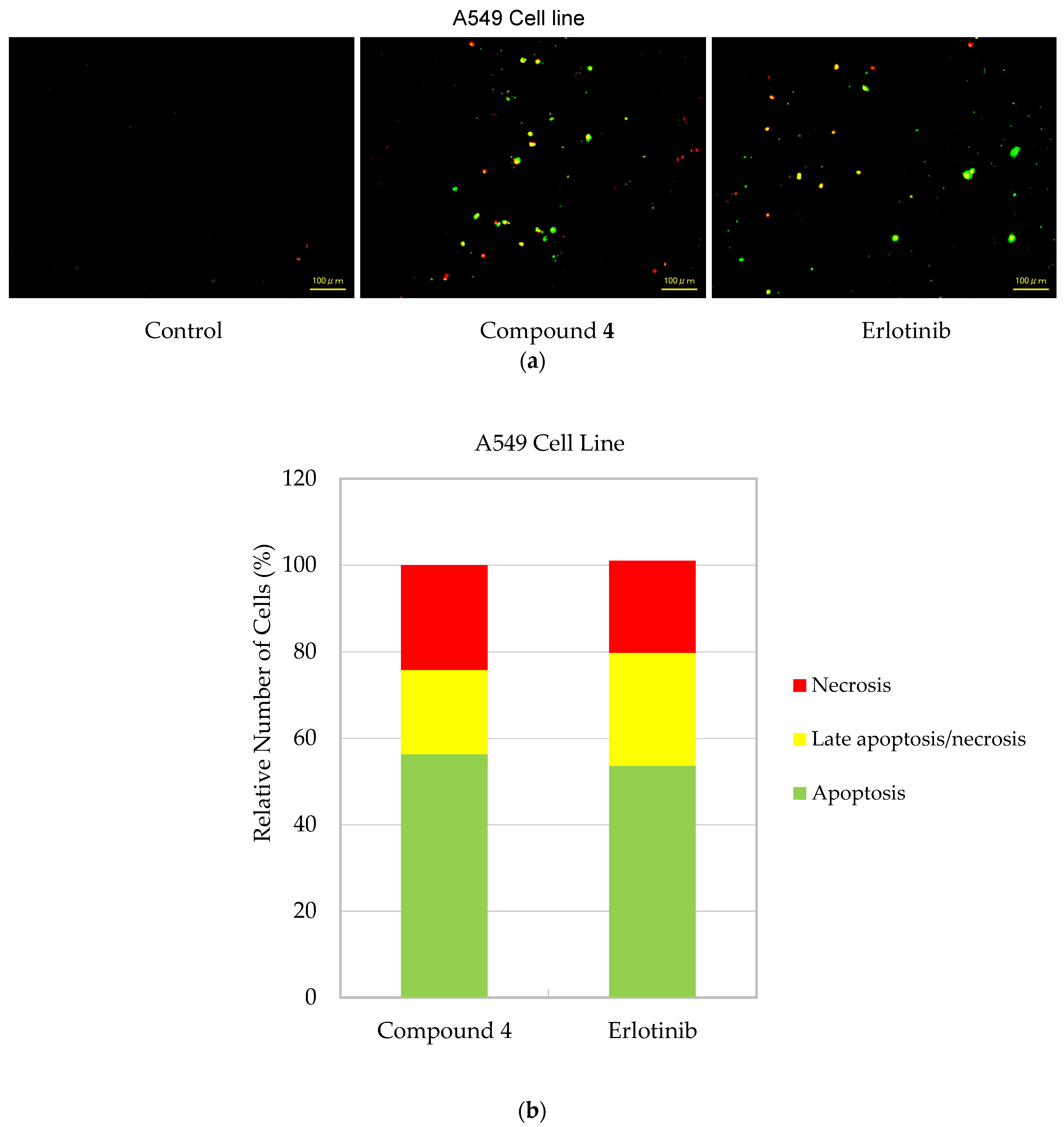
3.2.4. Detection of Cell Death
3.3. Molecular Docking
3.4. In Silico ADME Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Stewart, D.J.; Kurzrock, R. Targeted therapy in non-small-cell lung cancer—Is it becoming a reality? Nat. Rev. Clin. Oncol. 2010, 7, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. Molecular histology of lung cancer: From targets to treatments. Cancer Treat Rev. 2015, 41, 361–375. [Google Scholar] [CrossRef]
- Nascimento, A.V.; Bousbaa, H.; Ferreira, D.; Sarmento, B. Non-small cell lung carcinoma: An overview on targeted therapy. Curr. Drug Targets 2015, 16, 1448–1463. [Google Scholar] [CrossRef]
- Li, L.; Zhu, T.; Gao, Y.-F.; Zheng, W.; Wang, C.-J.; Xiao, L.; Huang, M.-S.; Yin, J.-Y.; Zhou, H.-H.; Liu, Z.-Q. Targeting DNA damage response in the radio(chemo)therapy of non-small cell lung cancer. Int. J. Mol. Sci. 2016, 17, 839. [Google Scholar] [CrossRef]
- Gyoba, J.; Shan, S.; Roa, W.; Bédard, E.L. Diagnosing lung cancers through examination of micro-RNA biomarkers in blood, plasma, serum and sputum: A review and summary of current literature. Int. J. Mol. Sci. 2016, 17, 494. [Google Scholar] [CrossRef] [Green Version]
- Kryczka, J.; Kryczka, J.; Czarnecka-Chrebelska, K.H.; Brzeziańska-Lasota, E. Molecular mechanisms of chemoresistance induced by cisplatin in NSCLC cancer therapy. Int. J. Mol. Sci. 2021, 22, 8885. [Google Scholar] [CrossRef] [PubMed]
- Iksen; Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/mTOR signaling pathway in lung cancer: An update regarding potential drugs and natural products. Molecules 2021, 26, 4100. [Google Scholar] [CrossRef]
- Singh, V.K.; Coumar, M.S. Chronic myeloid leukemia: Existing therapeutic options and strategies to overcome drug resistance. Mini-Rev. Med. Chem. 2019, 19, 333–345. [Google Scholar] [CrossRef]
- Ruiz-Ceja, K.A.; Chirino, Y.I. Current FDA-approved treatments for non-small cell lung cancer and potential biomarkers for its detection. Biomed. Pharmacother. 2017, 90, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Mustachio, L.M.; Roszik, J. Current targeted therapies for the fight against non-small cell lung cancer. Pharmaceuticals 2020, 13, 374. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, T.; Zhang, J. Natural tyrosine kinase inhibitors acting on the epidermal growth factor receptor: Their relevance for cancer therapy. Pharmacol. Res. 2020, 161, 105164. [Google Scholar] [CrossRef]
- Lee, D.H. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol. Ther. 2017, 174, 1–21. [Google Scholar] [CrossRef]
- Chen, L.; Fu, W.; Zheng, L.; Liu, Z.; Liang, G. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer. J. Med. Chem. 2018, 61, 4290–4300. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M mutation: A double role in lung cancer cell survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Le, T.; Gerber, D.E. Newer-generation EGFR inhibitors in lung cancer: How are they best used? Cancers 2019, 11, 366. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, I.; Planchard, D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front. Med. 2017, 3, 76. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Shareef, M.A.; Kumar, C.G. An overview on the synthetic and medicinal perspectives of indenopyrazoles. Eur. J. Med. Chem. 2019, 178, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Garikapati, K.R.; Setti, A.; Shaik, A.B.; Kanth Makani, V.K.; Shareef, M.A.; Rajpurohit, H.; Vangara, N.; Pal-Bhadra, M.; Kamal, A.; et al. Design, synthesis, in silico pharmacokinetics prediction and biological evaluation of 1,4-dihydroindeno[1,2-c]pyrazole chalcone as EGFR /Akt pathway inhibitors. Eur. J. Med. Chem. 2019, 163, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Garikapati, K.R.; Shaik, A.B.; Makani, V.K.K.; Rahim, A.; Shareef, M.A.; Reddy, V.G.; Pal-Bhadra, M.; Kamal, A.; Kumar, C.G. Design, synthesis and biological evaluation of 1, 4-dihydro indeno[1,2-c] pyrazole linked oxindole analogues as potential anticancer agents targeting tubulin and inducing p53 dependent apoptosis. Eur. J. Med. Chem. 2018, 144, 104–115. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Wang, J.-J.; Ji, Y.-T.; Zhao, G.-D.; Tang, L.-Q.; Zhang, C.-M.; Guo, X.-L.; Liu, Z.-P. Design, synthesis, and biological evaluation of 1-methyl-1,4-dihydroindeno[1,2-c]pyrazole analogues as potential anticancer agents targeting tubulin colchicine binding site. J. Med. Chem. 2016, 59, 5341–5355. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Ban, H.S.; Kawada, J.; Hirokawa, T.; Nakamura, H. Discovery of indenopyrazoles as EGFR and VEGFR-2 tyrosine kinase inhibitors by in silico high-throughput screening. Bioorg. Med. Chem. Lett. 2008, 18, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Claiborne, A.; Pyzytulinska, M.; Tao, Z.-F.; Stewart, K.D.; Kovar, P.; Chen, Z.; Credo, R.B.; Guan, R.; Merta, P.J.; et al. 1,4-Dihydroindeno[1,2-c]pyrazoles as potent checkpoint kinase 1 inhibitors: Extended exploration on phenyl ring substitutions and preliminary ADME/PK studies. Bioorg. Med. Chem. Lett. 2007, 17, 3618–3623. [Google Scholar] [CrossRef]
- Dinges, J.; Ashworth, K.L.; Akritopoulou-Zanze, I.; Arnold, L.D.; Baumeister, S.A.; Bousquet, P.F.; Cunha, G.A.; Davidsen, S.K.; Djuric, S.W.; Gracias, V.J.; et al. 1,4-Dihydroindeno[1,2-c]pyrazoles as novel multitargeted receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4266–4271. [Google Scholar] [CrossRef]
- Dinges, J.; Akritopoulou-Zanze, I.; Arnold, L.D.; Barlozzari, T.; Bousquet, P.F.; Cunha, G.A.; Ericsson, A.M.; Iwasaki, N.; Michaelides, M.R.; Ogawa, N.; et al. Hit-to-lead optimization of 1,4-dihydroindeno[1,2-c]pyrazoles as a novel class of KDR kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4371–4375. [Google Scholar] [CrossRef]
- Ho, C.Y.; Ludovici, D.W.; Maharoof, U.S.; Mei, J.; Sechler, J.L.; Tuman, R.W.; Strobel, E.D.; Andraka, L.; Yen, H.K.; Leo, G.; et al. (6,7-Dimethoxy-2,4-dihydroindeno[1,2-c]pyrazol-3-yl)phenylamines: Platelet-derived growth factor receptor tyrosine kinase inhibitors with broad antiproliferative activity against tumor cells. J. Med. Chem. 2005, 48, 8163–8173. [Google Scholar] [CrossRef]
- Nugiel, D.A.; Vidwans, A.; Etzkorn, A.M.; Rossi, K.A.; Benfield, P.A.; Burton, C.R.; Cox, S.; Doleniak, D.; Seitz, S.P. Synthesis and evaluation of indenopyrazoles as cyclin-dependent kinase inhibitors. 2. Probing the indeno ring substituent pattern. J. Med. Chem. 2002, 45, 5224–5232. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Samy, J.G.; Dutt, K.R.; Agrawal, U.K.; Yadav, B.S.; Vyas, S.; Kaur, R.; Yadav, G. Design, synthesis and antimycobacterial evaluation of novel 3-substituted-N-aryl-6,7-dimethoxy-3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide analogues. Bioorg. Med. Chem. Lett. 2011, 21, 4451–4453. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Samy, J.G.; Khalilullah, H.; Bakht, M.A.; Hassan, M.Z. Synthesis and antimycobacterial evaluation of 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide analogues. Eur. J. Med. Chem. 2011, 46, 5694–5697. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, A.; Gökbulut, S.; Sever, B.; Akalın Çiftçi, G.; Altıntop, M.D. Synthesis and evaluation of a new series of arylidene indanones as potential anticancer agents. Anti-Cancer Agents Med. Chem. 2018, 18, 1394–1404. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35 (Suppl.), S78–S103. [Google Scholar] [CrossRef]
- Goel, S.; Hidalgo, M.; Perez-Soler, R. EGFR inhibitor-mediated apoptosis in solid tumors. J. Exp. Ther. Oncol. 2007, 6, 305–320. [Google Scholar]
- Ciftci, H.I.; Radwan, M.O.; Ozturk, S.E.; Ulusoy, N.G.; Sozer, E.; Ellakwa, D.E.; Ocak, Z.; Can, M.; Ali, T.F.S.; Abd-Alla, H.I.; et al. Design, synthesis and biological evaluation of pentacyclic triterpene derivatives: Optimization of anti-ABL kinase activity. Molecules 2019, 24, 3535. [Google Scholar] [CrossRef] [Green Version]
- Bayrak, N.; Yıldırım, H.; Yıldız, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Ciftci, H.I.; Tuyun, A.F. A novel series of chlorinated plastoquinone analogs: Design, synthesis, and evaluation of anticancer activity. Chem. Biol. Drug Des. 2020, 95, 343–354. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Bayrak, N.; Yıldız, M.; Yıldırım, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Design, synthesis and investigation of the mechanism of action underlying anti-leukemic effects of the quinolinequinones as LY83583 analogs. Bioorg. Chem. 2021, 114, 105160. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Ellakwa, D.E.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Okiyama, Y.; et al. Antiproliferative S-trityl-L-cysteine -derived compounds as SIRT2 inhibitors: Repurposing and solubility enhancement. Molecules 2019, 24, 3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrak, N.; Ciftci, H.I.; Yıldız, M.; Yıldırım, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Structure based design, synthesis, and evaluation of anti-CML activity of the quinolinequinones as LY83583 analogs. Chem. Biol. Interact. 2021, 345, 109555. [Google Scholar] [CrossRef]
- Zeytün, E.; Altıntop, M.D.; Sever, B.; Özdemir, A.; Ellakwa, D.E.; Ocak, Z.; Ciftci, H.I.; Otsuka, M.; Fujita, M.; Radwan, M.O. A new series of antileukemic agents: Design, synthesis, in vitro and in silico evaluation of thiazole-based ABL1 kinase inhibitors. Anticancer Agents Med. Chem. 2021, 21, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H. Effects of glycyrrhetic acid on human chronic myelogenous leukemia cells. Turk. J. Pharm. Sci. 2020, 17, 49–55. [Google Scholar] [CrossRef]
- Sever, B.; Altıntop, M.D.; Özdemir, A.; Akalın Çiftçi, G.; Ellakwa, D.E.; Tateishi, H.; Radwan, M.O.; Ibrahim, M.A.A.; Otsuka, M.; Fujita, M.; et al. In vitro and in silico evaluation of anticancer activity of new indole-based 1,3,4-oxadiazoles as EGFR and COX-2 inhibitors. Molecules 2020, 25, 5190. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Radwan, M.O.; Sever, B.; Hamdy, A.K.; Emirdağ, S.; Ulusoy, N.G.; Sozer, E.; Can, M.; Yayli, N.; Araki, N.; et al. EGFR-targeted pentacyclic triterpene analogues for glioma therapy. Int. J. Mol. Sci. 2021, 22, 10945. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Can, M.; Ellakwa, D.E.; Suner, S.C.; Ibrahim, M.A.; Oral, A.; Sekeroglu, N.; Özalp, B.; Otsuka, M.; Fujita, M.; et al. Anticancer activity of Turkish marine extracts: A purple sponge extract induces apoptosis with multitarget kinase inhibition activity. Invest. New Drugs 2020, 38, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Ciftci, H.I.; Radwan, M.O.; Sever, B.; Kaplancıklı, Z.A.; Ali, T.F.S.; Koga, R.; Fujita, M.; Otsuka, M.; Özdemir, A. Design, synthesis, and biological evaluation of novel 1,3,4-thiadiazole derivatives as potential antitumor agents against chronic myelogenous leukemia: Striking effect of nitrothiazole moiety. Molecules 2018, 23, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; et al. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci. Rep. 2017, 7, 8957. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | IC50 (µM) | SI 1 | ||
|---|---|---|---|---|---|
| A549 Cells | K562 Cells | PBMCs | |||
| 1 | H | 7.99 ± 1.84 | 3.49 ± 0.92 | 95.72 ± 11.68 | 27.43 |
| 2 | F | >10 | |||
| 3 | Cl | >10 | |||
| 4 | Br | 6.13 ± 1.42 | 2.65 ± 0.75 | 19.56 ± 4.25 | 7.38 |
| 5 | CN | >10 | |||
| 6 | SO2CH3 | >10 | |||
| 7 | CH3 | 8.67 ± 2.18 | 2.76 ± 0.83 | >100 | >36.23 |
| Erlotinib | - | 19.67 ± 3.15 | 29.62 ± 5.26 | 44.33 ± 7.84 | 1.50 |
| Compound | QPlogBB * (−3–1.2) | CNS ** (−2 to 2) | Human Oral Absorption% *** | Rule of Five **** | Rule of Three ***** |
|---|---|---|---|---|---|
| 1 | 0.478 | 2 | 100 | 1 | 1 |
| 2 | 0.592 | 2 | 100 | 1 | 1 |
| 3 | 0.650 | 2 | 100 | 1 | 1 |
| 4 | 0.663 | 2 | 100 | 2 | 1 |
| 5 | −0.388 | 0 | 100 | 1 | 1 |
| 6 | −0.495 | 0 | 94.085 | 2 | 1 |
| 7 | 0.472 | 2 | 100 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özdemir, A.; Ciftci, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Altıntop, M.D. A New Series of Indeno[1,2-c]pyrazoles as EGFR TK Inhibitors for NSCLC Therapy. Molecules 2022, 27, 485. https://doi.org/10.3390/molecules27020485
Özdemir A, Ciftci H, Sever B, Tateishi H, Otsuka M, Fujita M, Altıntop MD. A New Series of Indeno[1,2-c]pyrazoles as EGFR TK Inhibitors for NSCLC Therapy. Molecules. 2022; 27(2):485. https://doi.org/10.3390/molecules27020485
Chicago/Turabian StyleÖzdemir, Ahmet, Halilibrahim Ciftci, Belgin Sever, Hiroshi Tateishi, Masami Otsuka, Mikako Fujita, and Mehlika Dilek Altıntop. 2022. "A New Series of Indeno[1,2-c]pyrazoles as EGFR TK Inhibitors for NSCLC Therapy" Molecules 27, no. 2: 485. https://doi.org/10.3390/molecules27020485