
Copolymers Derived from Two Active Esters: Synthesis, Characterization, Thermal Properties, and Reactivity in Post-Modification

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
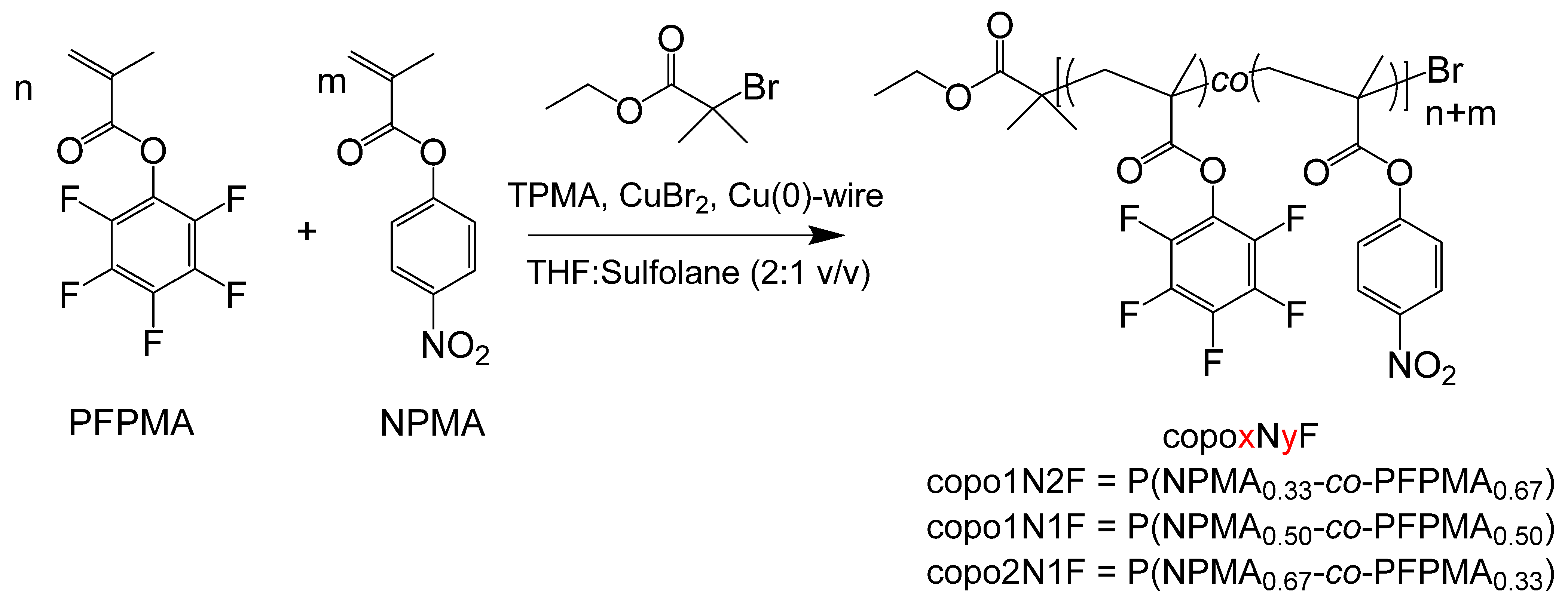
2.1. Synthesis of Copolymers of PFPMA and NPMA
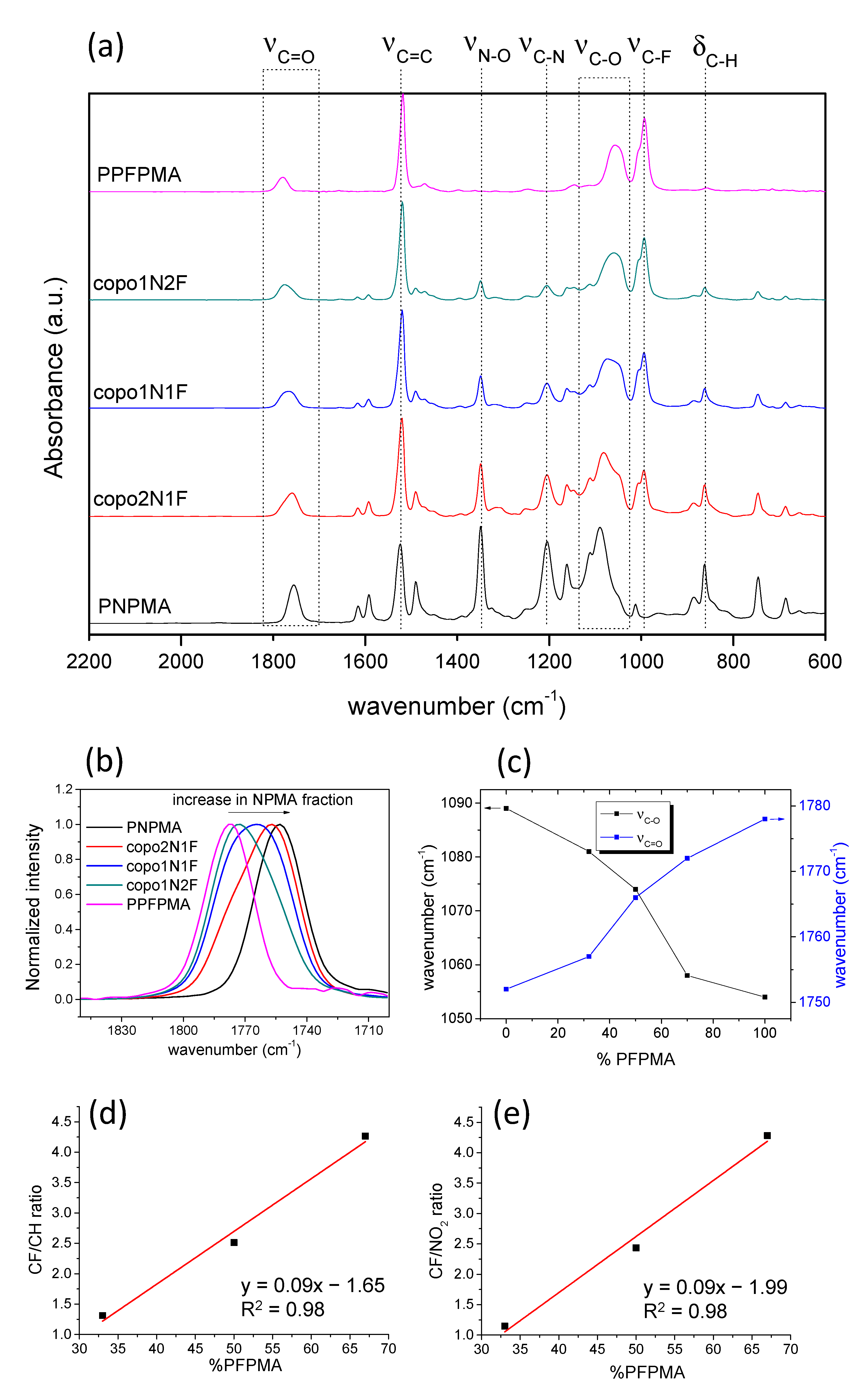
2.2. FTIR Analysis: Chemical Environment Analysis
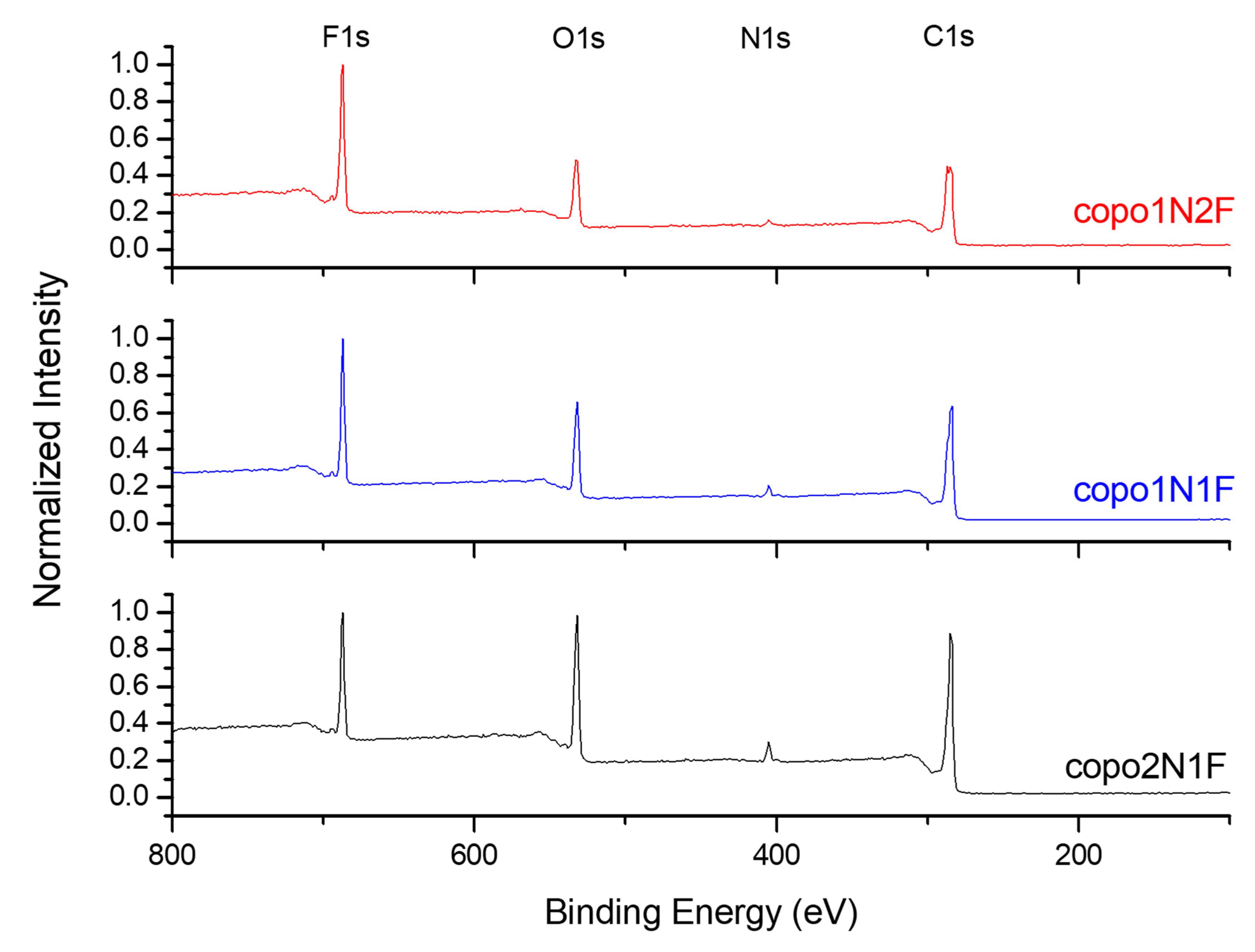
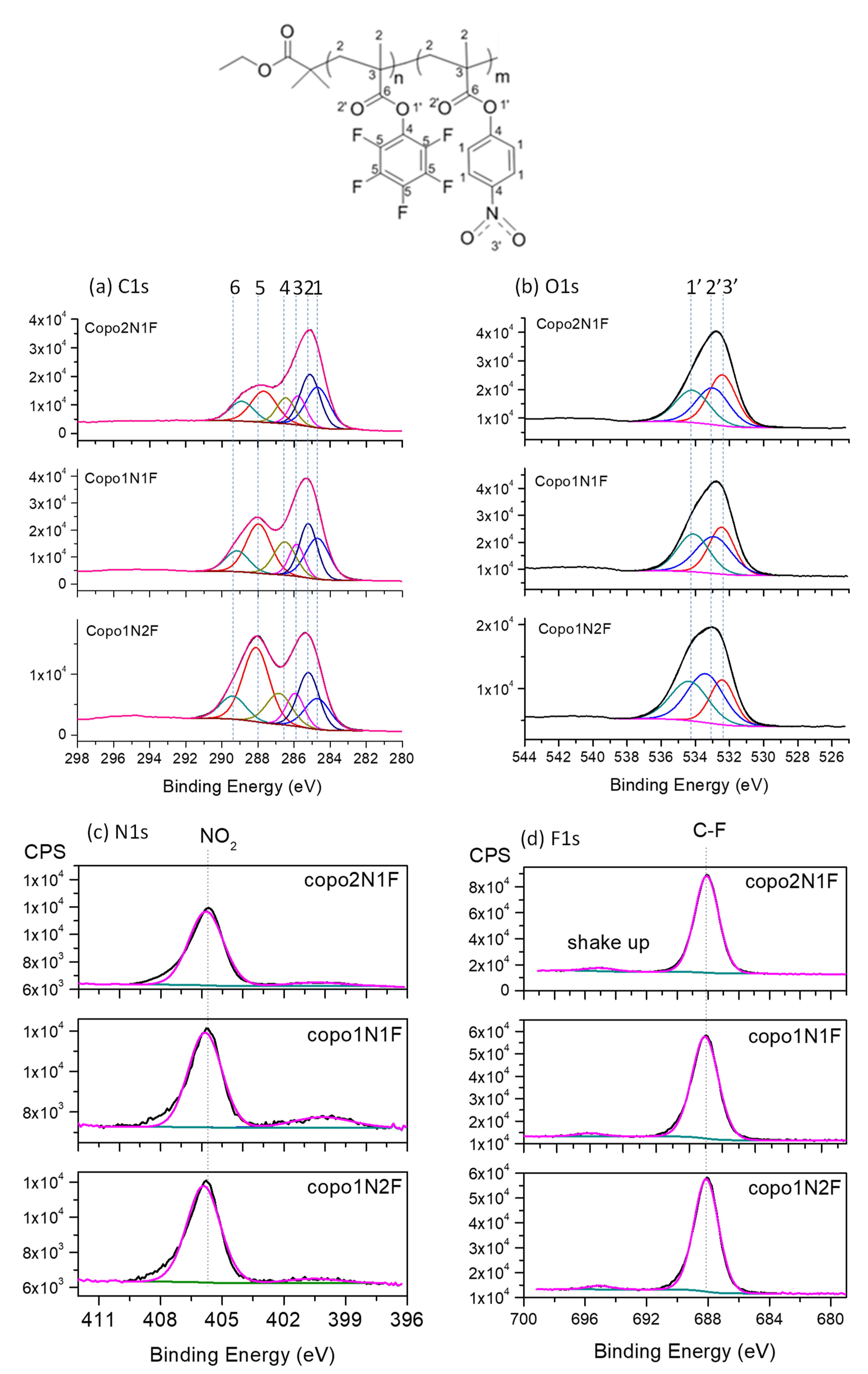
2.3. X-ray Photon Spectroscopy: Details on Composition of Copolymers
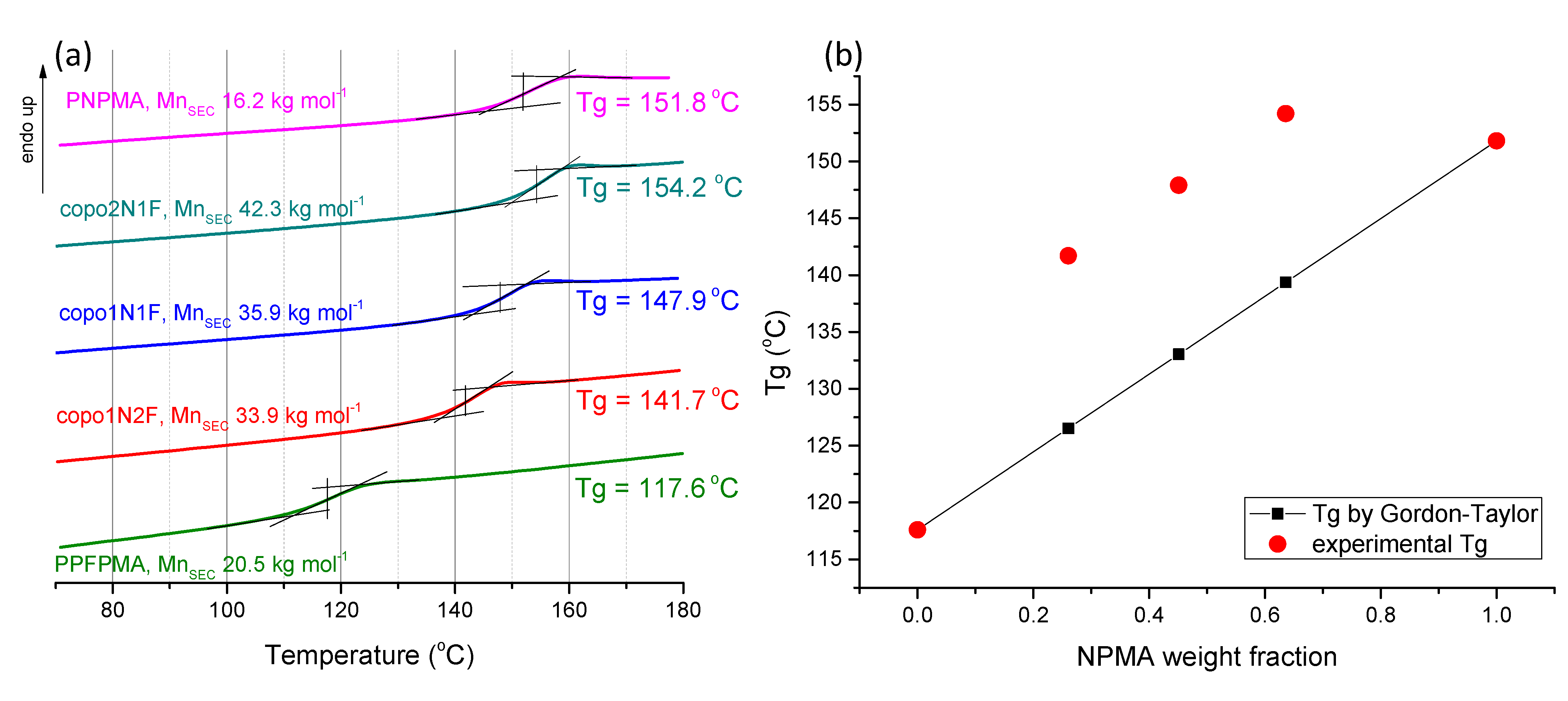
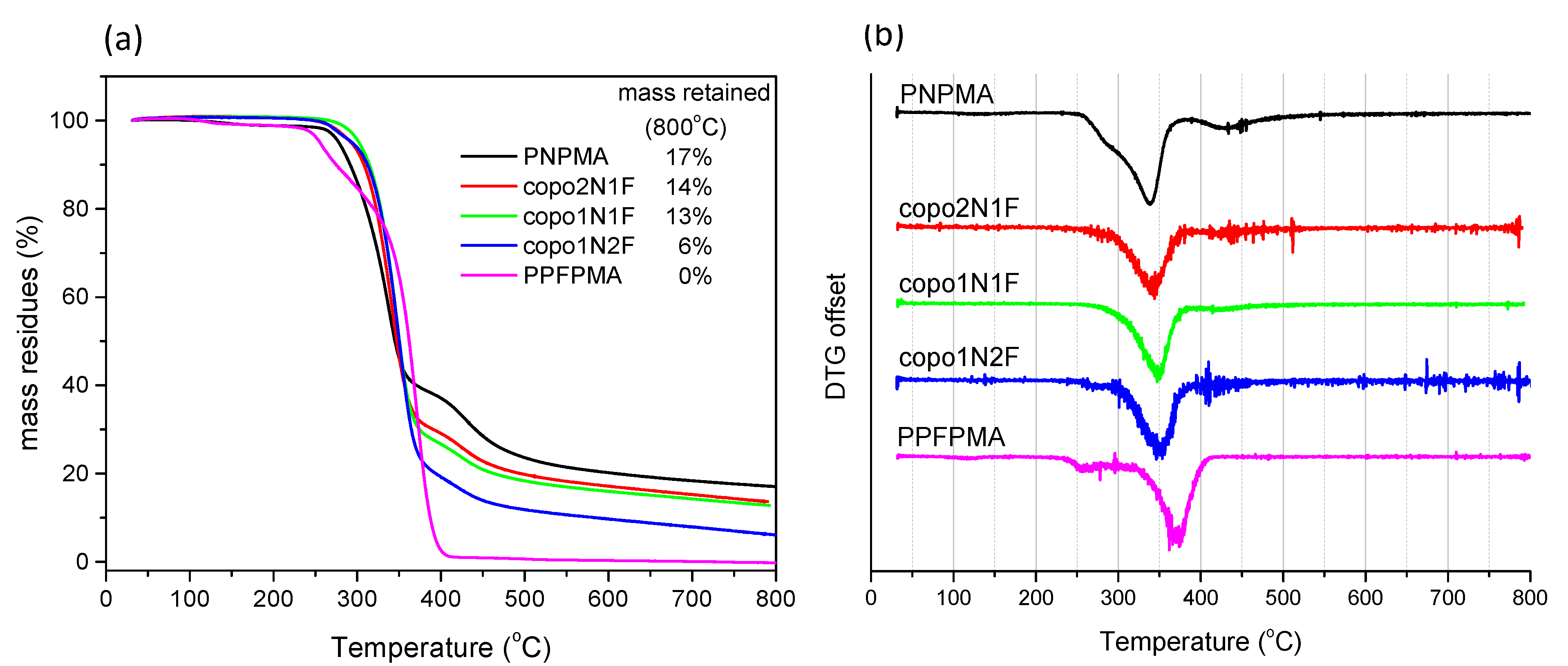
2.4. Thermal Properties of Copolymers
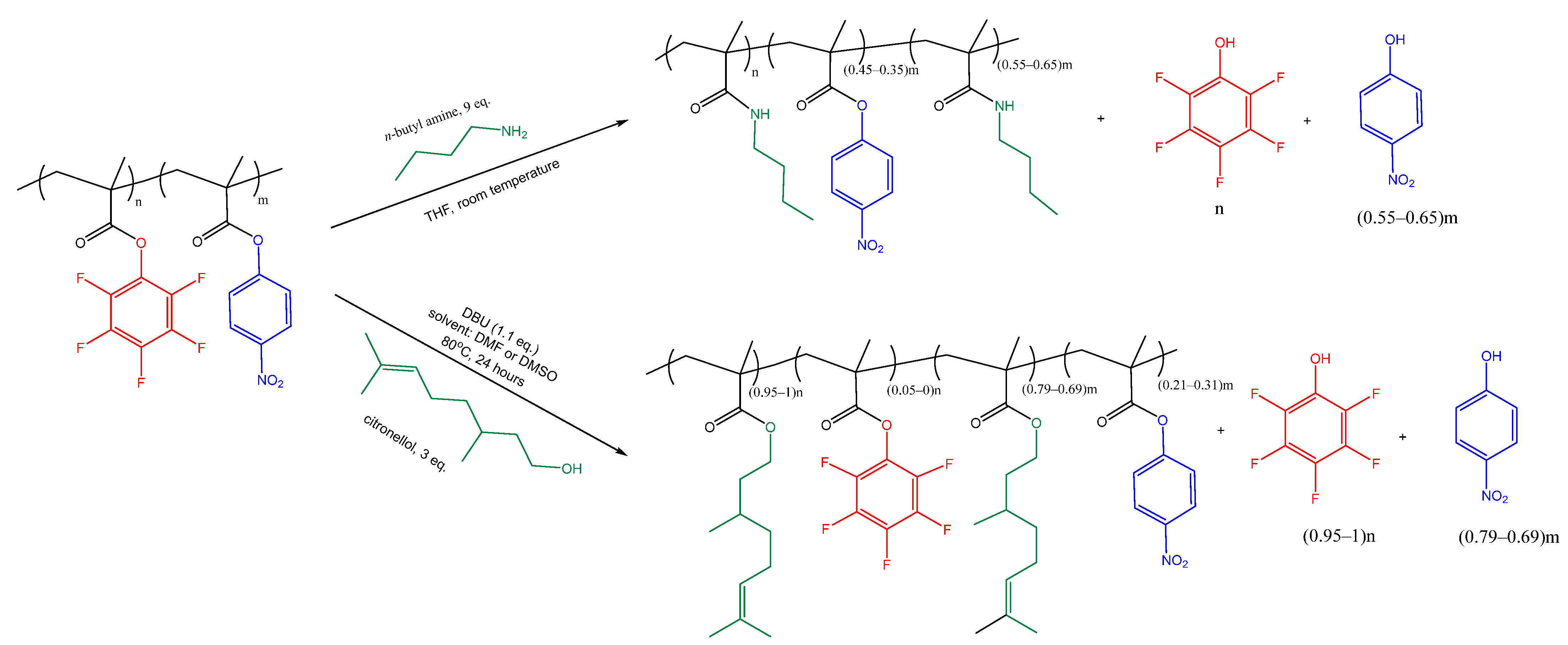
2.5. Post-Modification of Copolymers
3. Materials and Methods
3.1. Materials
3.2. Characterization
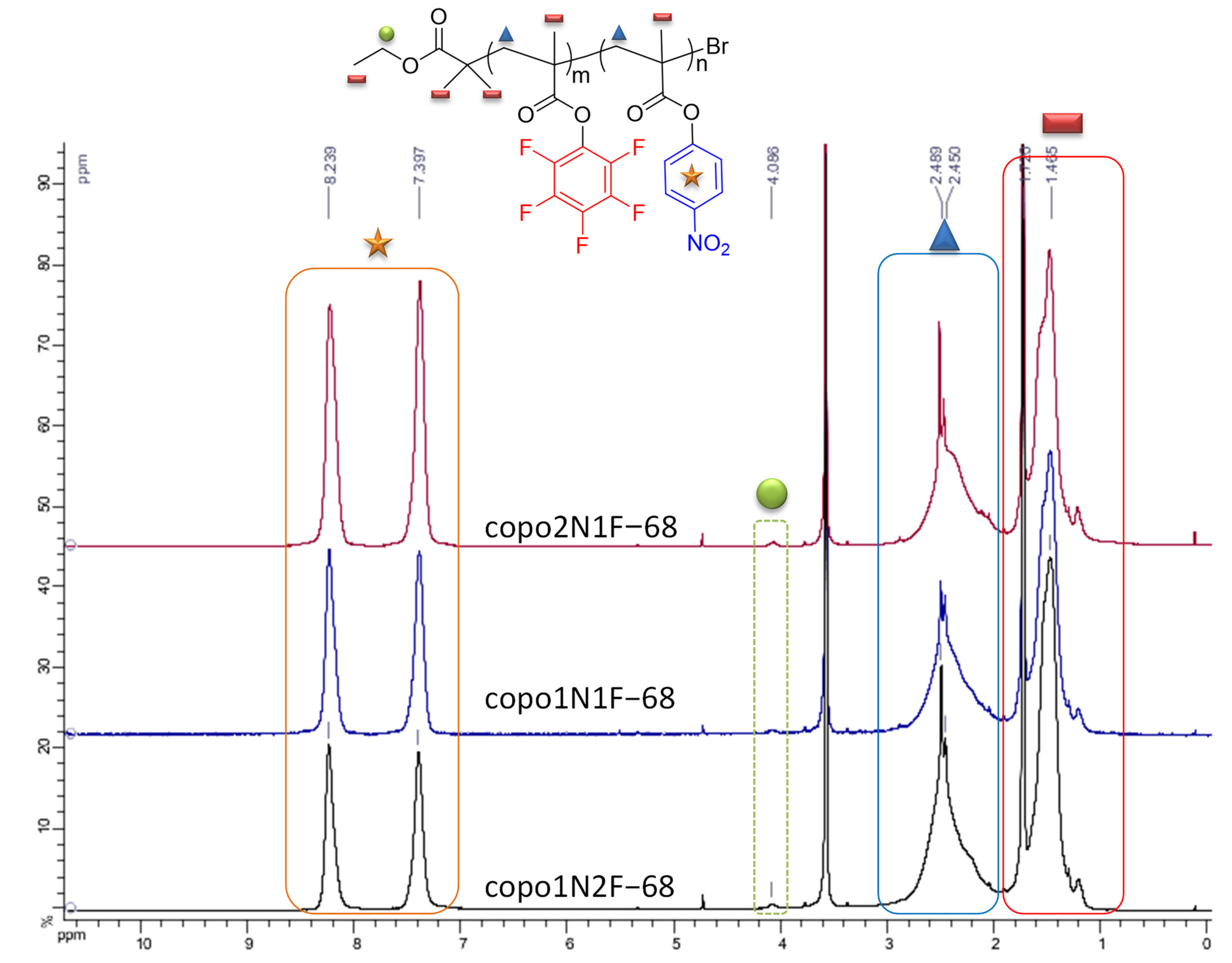
3.2.1. NMR
3.2.2. SEC
3.2.3. X-ray Photoelectron Spectroscopy
3.2.4. Elemental Analysis
3.2.5. Attenuated Total Reflectance (ATR) FT-IR
3.2.6. Thermal Analysis
3.3. Method
3.3.1. Synthesis of PFPMA
3.3.2. Synthesis of NPMA
3.3.3. Cu(0)-mediated RDRP of PFPMA and NPMA
3.3.4. Post-Modification of Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gauthier, M.A.; Gibson, M.I.; Klok, H.-A. Synthesis of Functional Polymers by Post-Polymerization Modification. Angew. Chem. Int. Ed. 2009, 48, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Bettelli, A.; Feré, A. High Polymers of Acrylic and Methacrylic Esters of N-Hydroxysuccinimide as Polyacrylamide and Polymethacrylamide Precursors. Polymer 1972, 13, 462–464. [Google Scholar] [CrossRef]
- Erout, M.-N.; Troesch, A.; Pichot, C.; Cros, P. Preparation of Conjugates between Oligonucleotides and N-Vinylpyrrolidone/N-Acryloxysuccinimide Copolymers and Applications in Nucleic Acid Assays to Improve Sensitivity. Bioconjugate Chem. 1996, 7, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.; Mruk, R.; Zentel, R.; Théato, P. Synthesis of Pentafluorophenyl (Meth) Acrylate Polymers: New Precursor Polymers for the Synthesis of Multifunctional Materials. Eur. Polym. J. 2005, 41, 1569–1575. [Google Scholar] [CrossRef]
- Das, A.; Theato, P. Multifaceted Synthetic Route to Functional Polyacrylates by Transesterification of Poly (Pentafluorophenyl Acrylates). Macromolecules 2015, 48, 8695–8707. [Google Scholar] [CrossRef]
- Arnold, R.M.; McNitt, C.D.; Popik, V.V.; Locklin, J. Direct Grafting of Poly (Pentafluorophenyl Acrylate) onto Oxides: Versatile Substrates for Reactive Microcapillary Printing and Self-Sorting Modification. Chem. Commun. 2014, 50, 5307–5309. [Google Scholar] [CrossRef]
- Ye, L.; Cormack, P.A.G.; Mosbach, K. Molecular Imprinting on Microgel Spheres. Anal. Chim. Acta 2001, 435, 187–196. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Pan, C. Synthesis of Block Copoly (Styrene-b-p-Nitrophenyl Methacrylate) and Its Derivatives by Atom Transfer Radical Polymerization. Macromolecules 1999, 32, 8301–8305. [Google Scholar] [CrossRef]
- Benaglia, M.; Alberti, A.; Giorgini, L.; Magnoni, F.; Tozzi, S. Poly (Glycidyl Methacrylate): A Highly Versatile Polymeric Building Block for Post-Polymerization Modifications. Polym. Chem. 2012, 4, 124–132. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-Polymerization Modification Reactions of Poly(Glycidyl Methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef]
- Kocak, G.; Solmaz, G.; Tuncer, C.; Bütün, V. Modification of Glycidyl Methacrylate Based Block Copolymers and Their Aqueous Solution Behaviours. Eur. Polym. J. 2019, 110, 364–377. [Google Scholar] [CrossRef]
- Le Gars, M.; Bras, J.; Salmi-Mani, H.; Ji, M.; Dragoe, D.; Faraj, H.; Domenek, S.; Belgacem, N.; Roger, P. Polymerization of Glycidyl Methacrylate from the Surface of Cellulose Nanocrystals for the Elaboration of PLA-Based Nanocomposites. Carbohydr. Polym. 2020, 234, 115899. [Google Scholar] [CrossRef]
- Molle, E.; Mutlu, H.; Theato, P. Synthesis and Post-Polymerization Modification of Poly (N-(4-Vinylphenyl)Sulfonamide)s. Macromol. Rapid Commun. 2021, 42, 2100063. [Google Scholar] [CrossRef]
- Pinyakit, Y.; Palaga, T.; Kiatkamjornwong, S.; Hoven, V.P. Sequential Post-Polymerization Modification of a Pentafluorophenyl Ester-Containing Homopolymer: A Convenient Route to Effective PH-Responsive Nanocarriers for Anticancer Drugs. J. Mater. Chem. B 2020, 8, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, F.; Meijer, E.W.; Palmans, A.R.A. Amphiphilic Polymeric Nanoparticles for Photoredox Catalysis in Water. Chem.—A Eur. J. 2020, 26, 10355–10361. [Google Scholar] [CrossRef]
- Nuhn, L.; Hartmann, S.; Palitzsch, B.; Gerlitzki, B.; Schmitt, E.; Zentel, R.; Kunz, H. Water-Soluble Polymers Coupled with Glycopeptide Antigens and T-Cell Epitopes as Potential Antitumor Vaccines. Angew. Chem. Int. Ed. 2013, 52, 10652–10656. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Shang, J.; Theato, P. Preparation of Dual Stimuli-Responsive Block Copolymers Based on Different Activated Esters with Distinct Reactivities. Eur. Polym. J. 2015, 69, 523–531. [Google Scholar] [CrossRef]
- Varadharajan, D.; Delaittre, G. Accessing Libraries of Bifunctional Block Copolymers Using Two Distinct Pentafluorophenyl Moieties. Polym. Chem. 2016, 7, 7488–7499. [Google Scholar] [CrossRef]
- Nguyen, T.P.T.; Barroca-Aubry, N.; Costa, L.; Bourdreux, Y.; Doisneau, G.; Roger, P. Cu(0)-Mediated RDRP as New Alternative for Controlled Synthesis of Poly (Pentafluorophenyl Methacrylate). Polymer 2022, 251, 124924. [Google Scholar] [CrossRef]
- Anastasaki, A.; Nikolaou, V.; Haddleton, D.M. Cu(0)-Mediated Living Radical Polymerization: Recent Highlights and Applications; a Perspective. Polym. Chem. 2016, 7, 1002–1026. [Google Scholar] [CrossRef]
- Zhang, T.; Du, Y.; Kalbacova, J.; Schubel, R.; Rodriguez, R.D.; Chen, T.; Zahn, D.R.T.; Jordan, R. Wafer-Scale Synthesis of Defined Polymer Brushes under Ambient Conditions. Polym. Chem. 2015, 6, 8176–8183. [Google Scholar] [CrossRef] [Green Version]
- Bach, L.G.; Islam, M.R.; Lee, D.C.; Lim, K.T. Poly (Glycidyl Methacrylate) Grafted CdSe Quantum Dots by Surface-Initiated Atom Transfer Radical Polymerization: Novel Synthesis, Characterization, Properties, and Cytotoxicity Studies. Appl. Surf. Sci. 2013, 283, 546–553. [Google Scholar] [CrossRef]
- Nguyen, T.P.T.; Barroca-Aubry, N.; Dragoe, D.; Mazerat, S.; Brisset, F.; Herry, J.-M.; Roger, P. Facile and Efficient Cu(0)-Mediated Radical Polymerisation of Pentafluorophenyl Methacrylate Grafting from Poly (Ethylene Terephthalate) Film. Eur. Polym. J. 2019, 116, 497–507. [Google Scholar] [CrossRef]
- Maaz, M.; Elzein, T.; Bejjani, A.; Barroca-Aubry, N.; Lepoittevin, B.; Dragoe, D.; Mazerat, S.; Nsouli, B.; Roger, P. Surface Initiated Supplemental Activator and Reducing Agent Atom Transfer Radical Polymerization (SI-SARA-ATRP) of 4-Vinylpyridine on Poly (Ethylene Terephthalate). J. Colloid Interface Sci. 2017, 500, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Maaz, M.; Elzein, T.; Barroca-Aubry, N.; Simoni, E.; Costa, L.; Nsouli, B.; Roger, P. New Insights on Uranium Recovery from Seawater and Aqueous Media. Appl. Mater. Today 2020, 18, 100461. [Google Scholar] [CrossRef]
- Boyer, C.; Corrigan, N.A.; Jung, K.; Nguyen, D.; Nguyen, T.-K.; Adnan, N.N.M.; Oliver, S.; Shanmugam, S.; Yeow, J. Copper-Mediated Living Radical Polymerization (Atom Transfer Radical Polymerization and Copper(0) Mediated Polymerization): From Fundamentals to Bioapplications. Chem. Rev. 2016, 116, 1803–1949. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Hori, N.; Kajiyama, M.; Takemura, A. Graft Modification of Methyl Acrylate onto Chicken Feather via Surface Initiated Cu(0)-Mediated Reversible-Deactivation Radical Polymerization. J. Appl. Polym. Sci. 2019, 136, 48246. [Google Scholar] [CrossRef]
- Alex, J.; Ulbrich, J.; Rosales-Guzmán, M.; Weber, C.; Schubert, U.S.; Guerrero-Sanchez, C. Kinetic Investigations on Homo- and Co-Polymerizations of Pentafluorophenyl (Meth)Acrylates. Eur. Polym. J. 2021, 143, 110175. [Google Scholar] [CrossRef]
- Thamizharasi, S.; Gnanasundaram, P.; Balasubramanian, S. Copolymers Derived from 4-Nitrophenyl Methacrylate (Npma) and Methyl-Methacrylate (MMA)-Synthesis, Characterization, and Reactivity Ratios. J. Macromol. Sci. Part A 1998, 35, 1835–1852. [Google Scholar] [CrossRef]
- Tagaya, A.; Harada, T.; Koike, K.; Koike, Y.; Okamoto, Y.; Teng, H.; Yang, L. Improvement of the Physical Properties of Poly (Methyl Methacrylate) by Copolymerization with Pentafluorophenyl Methacrylate. J. Appl. Polym. Sci. 2007, 106, 4219–4224. [Google Scholar] [CrossRef]
- Teng, H.; Yang, L.; Mikes, F.; Koike, Y.; Okamoto, Y. Property Modification of Poly (Methyl Methacrylate) through Copolymerization with Fluorinated Aryl Methacrylate Monomers. Polym. Adv. Technol. 2007, 18, 453–457. [Google Scholar] [CrossRef]
- Thamizharasi, S.; Gnanasundaram, P.; Reddy, B.S.R. Copolymerization of 4-nitrophenyl acrylate with glycidyl methacrylate: Synthesis, characterization, and reactivity ratios. J. Appl. Polym. Sci. 1997, 65, 1285–1291. [Google Scholar] [CrossRef]
- Soltanmorad, R.; Nas, M.H. Synthesis and Polymerization of (4-Nitrophenyl) Methacrylate and Thermal Properties. Cumhur. Sci. J. 2015, 36, 10. [Google Scholar]
- Koike, K.; Kado, T.; Satoh, Z.; Okamoto, Y.; Koike, Y. Optical and Thermal Properties of Methyl Methacrylate and Pentafluorophenyl Methacrylate Copolymer: Design of Copolymers for Low-Loss Optical Fibers for Gigabit in-Home Communications. Polymer 2010, 51, 1377–1385. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Adams, H.; Jimenez Blanco, J.-L.; Chessari, G.; Hunter, C.A.; Low, C.M.R.; Sanderson, J.M.; Vinter, J.G. Quantitative Determination of Intermolecular Interactions with Fluorinated Aromatic Rings. Chem.—A Eur. J. 2001, 7, 3494–3503. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | fPFPMA a | FPFPMA b | t (h) | Conversion (%) | Mn theo (g.mol−1) | SEC c | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mn SEC | Mw SEC | Đ | ||||||||
| PFPMA | NPMA | g.mol−1 | ||||||||
| 1 | Copo1N2F-19 | 0.67 | 0.66 | 19 | 69 | 71 | 16,900 | 38,900 | 58,000 | 1.49 |
| 2 | Copo1N1F-19 | 0.50 | 0.54 | 19 | 65 | 55 | 14,100 | 28,900 | 38,300 | 1.33 |
| 3 | Copo2N1F-19 | 0.33 | 0.32 | 19 | 64 | 68 | 14,800 | 42,100 | 62,700 | 1.49 |
| 4 | Copo1N2F-68 | 0.67 | 0.70 | 68 | 89 | 78 | 21,100 | 33,900 | 42,300 | 1.43 |
| 5 | Copo1N1F-68 | 0.50 | 0.50 | 68 | 98 | 98 | 22,700 | 35,900 | 61,300 | 1.71 |
| 6 | Copo2N1F-68 | 0.33 | 0.32 | 68 | 92 | 96 | 20,900 | 42,300 | 62,600 | 1.48 |
| 7 | Copo1N1F-M * | 0.50 | 0.51 | 20 | 80 | 76 | 20,200 | 17,100 d | 44,300 d | 2.59 d |
| 8 | PNPMA ** | 0 | 0 | 19 | - | 98 | 20,500 | 13,500 | 24,300 | 1.80 |
| 9 | PPFPMA *** | 1 | 1 | 19 | 90 | - | 22,900 | 35,500 d | 37,400 d | 1.05 d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.P.T.; Barroca-Aubry, N.; Aymes-Chodur, C.; Dragoe, D.; Pembouong, G.; Roger, P. Copolymers Derived from Two Active Esters: Synthesis, Characterization, Thermal Properties, and Reactivity in Post-Modification. Molecules 2022, 27, 6827. https://doi.org/10.3390/molecules27206827
Nguyen TPT, Barroca-Aubry N, Aymes-Chodur C, Dragoe D, Pembouong G, Roger P. Copolymers Derived from Two Active Esters: Synthesis, Characterization, Thermal Properties, and Reactivity in Post-Modification. Molecules. 2022; 27(20):6827. https://doi.org/10.3390/molecules27206827
Chicago/Turabian StyleNguyen, Thi Phuong Thu, Nadine Barroca-Aubry, Caroline Aymes-Chodur, Diana Dragoe, Gaëlle Pembouong, and Philippe Roger. 2022. "Copolymers Derived from Two Active Esters: Synthesis, Characterization, Thermal Properties, and Reactivity in Post-Modification" Molecules 27, no. 20: 6827. https://doi.org/10.3390/molecules27206827