
Chiral Bis(tetrathiafulvalene)-1,2-cyclohexane-diamides

Abstract
:
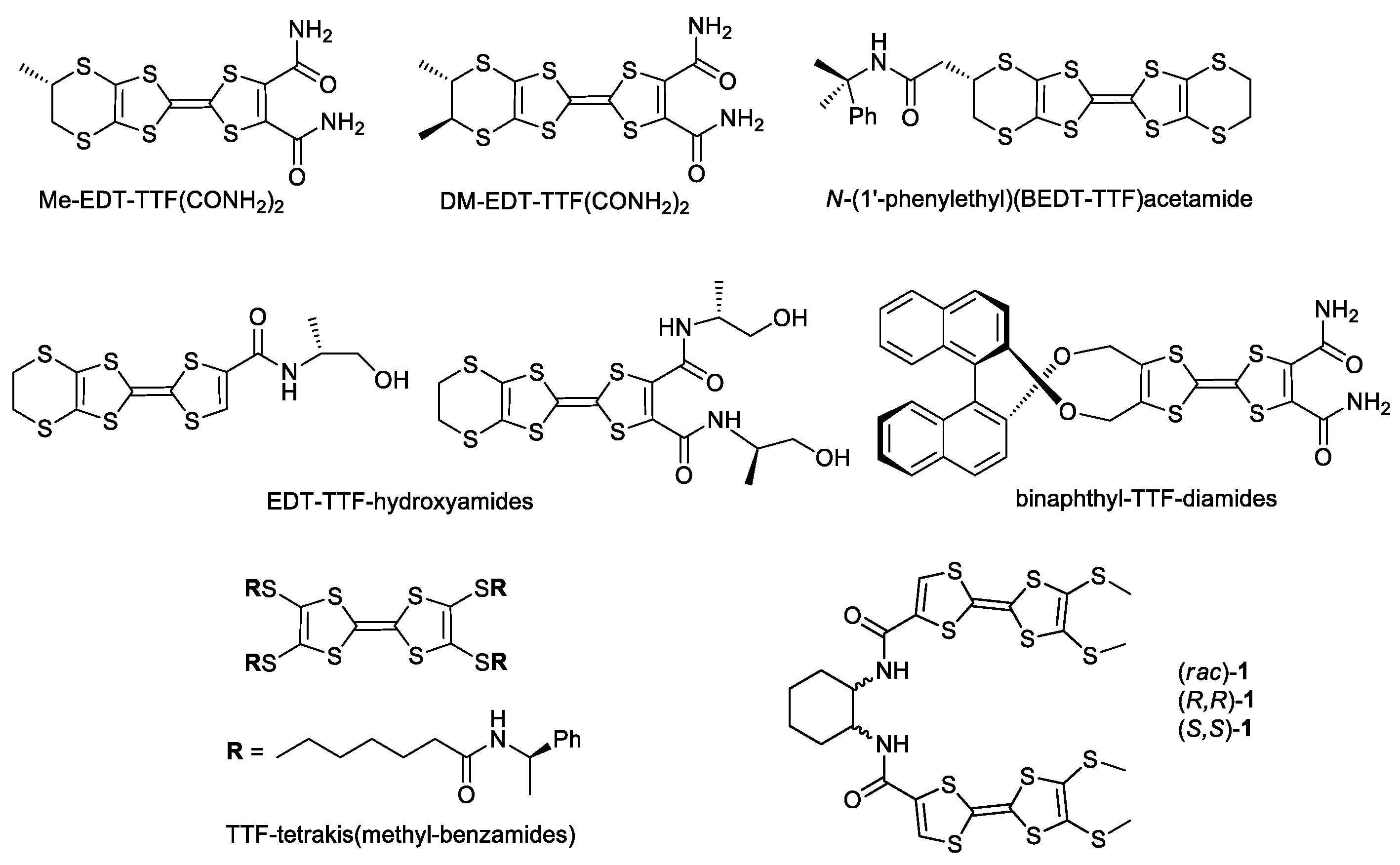
1. Introduction
2. Results and Discussion
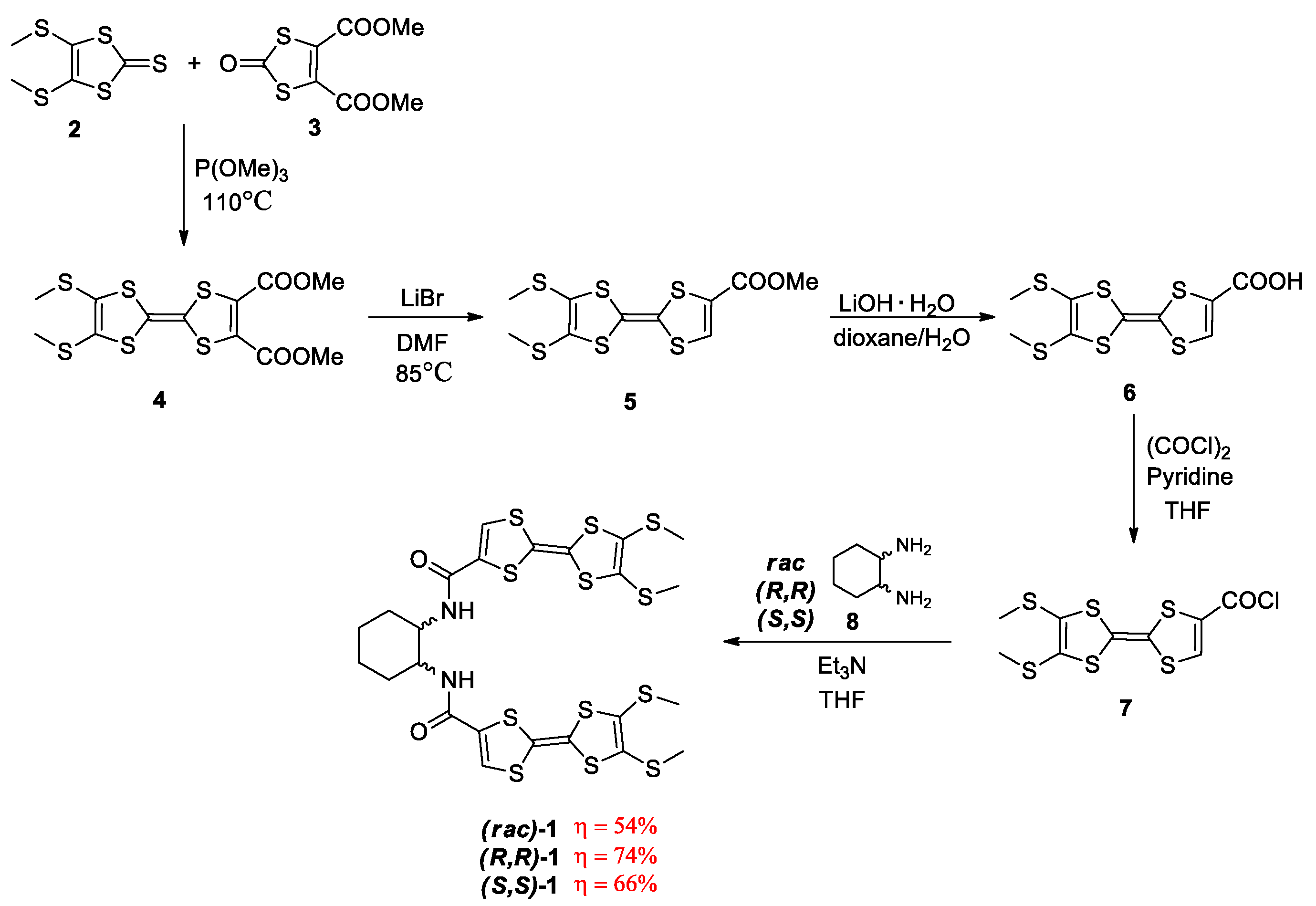
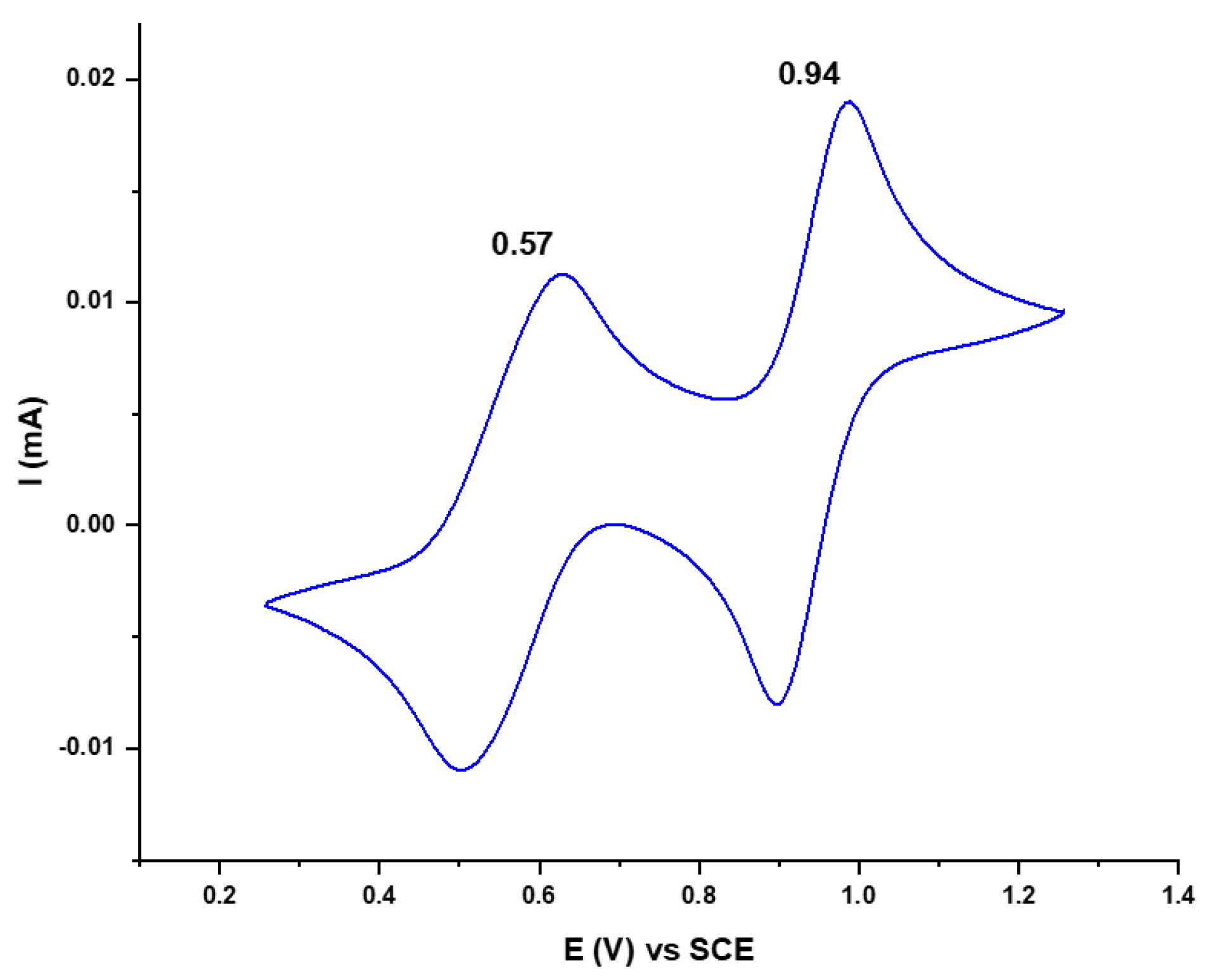
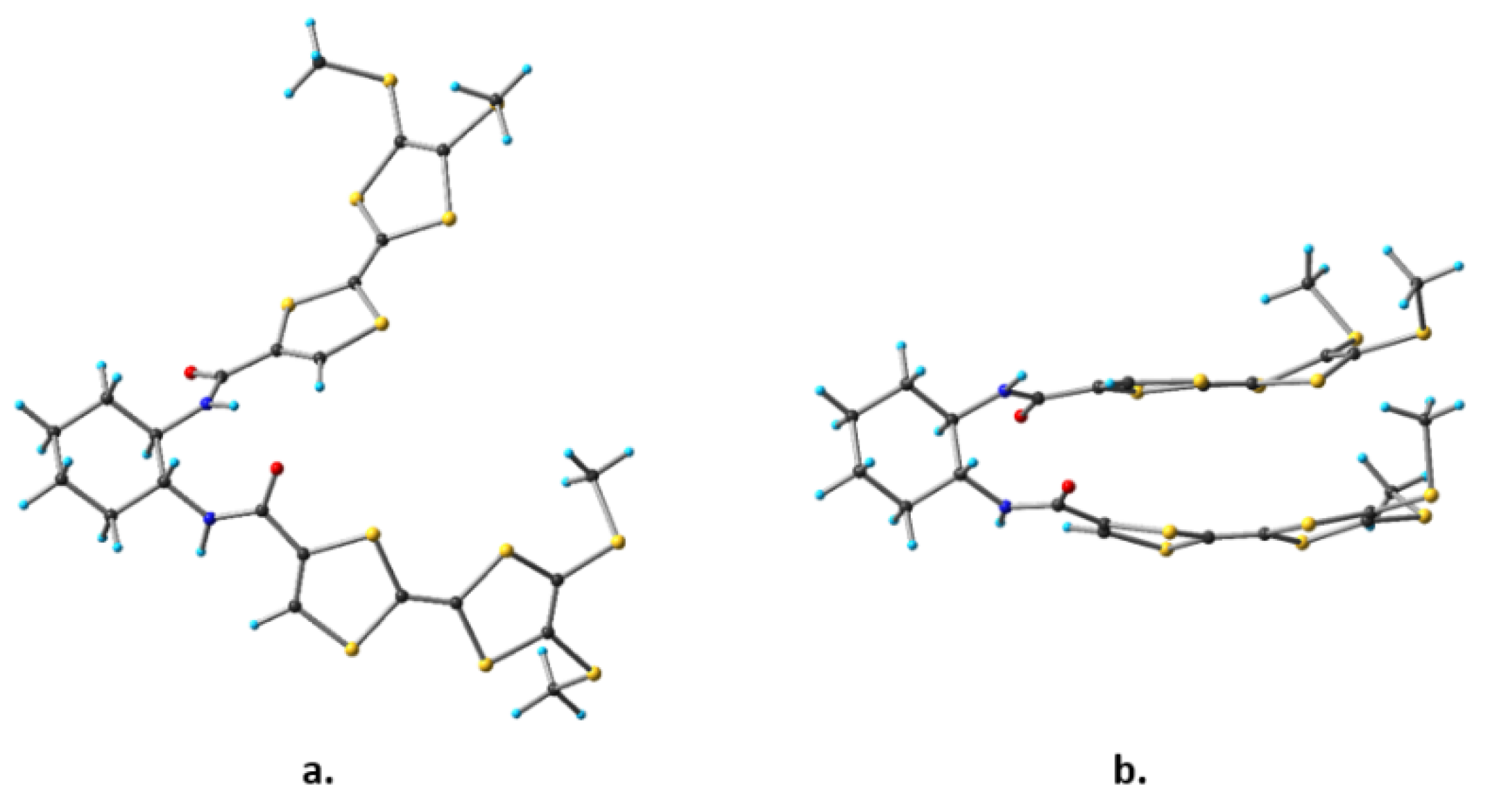
2.1. Synthesis and Characterization of Racemic and Enantiopure Donor 1
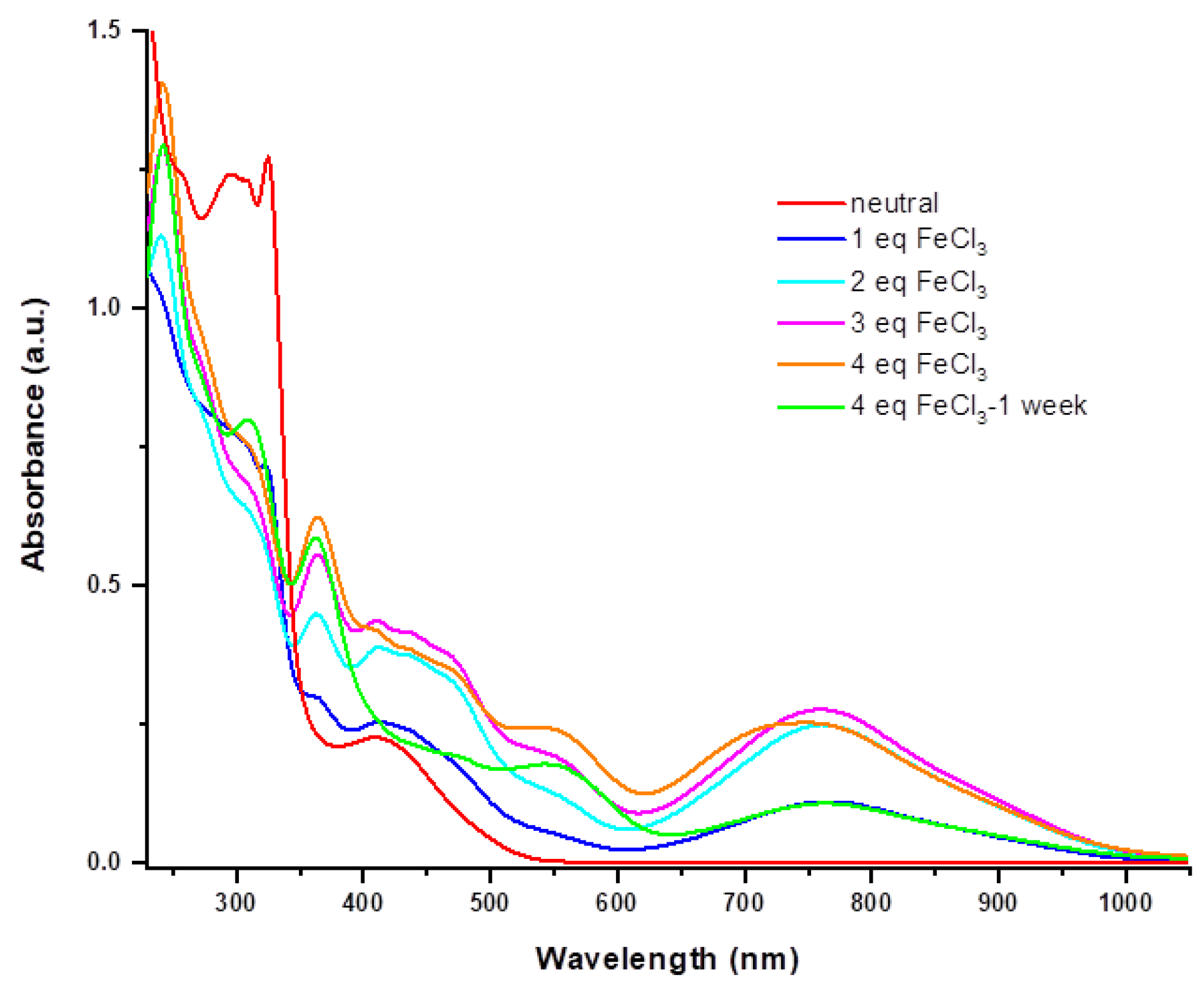
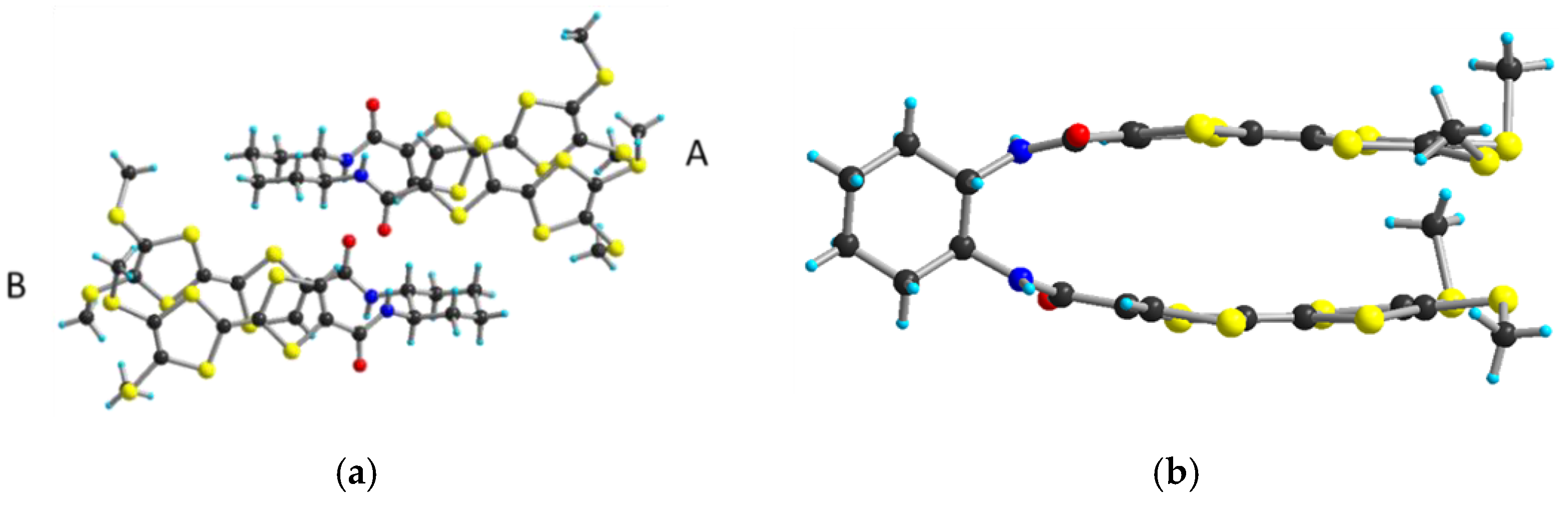
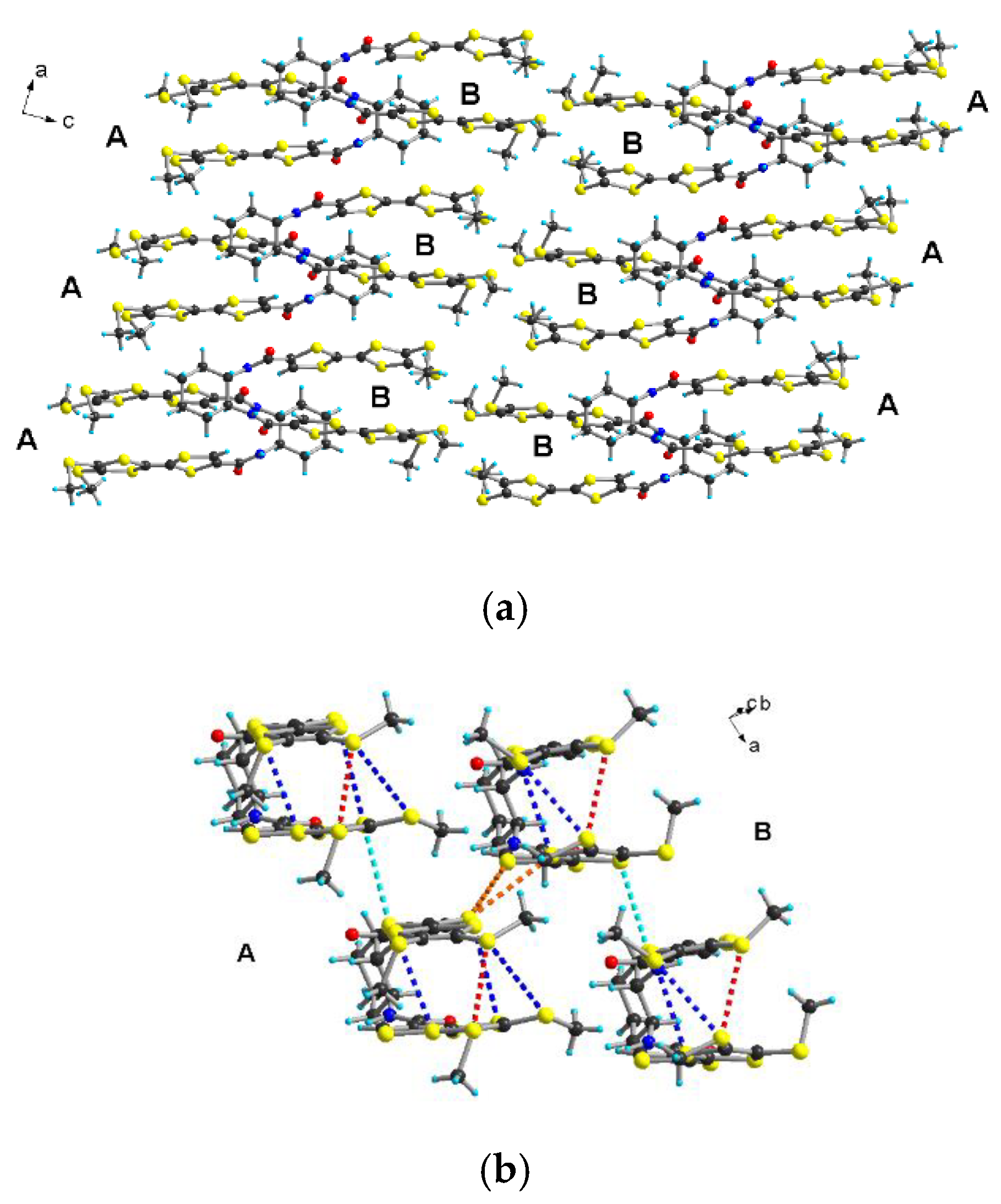
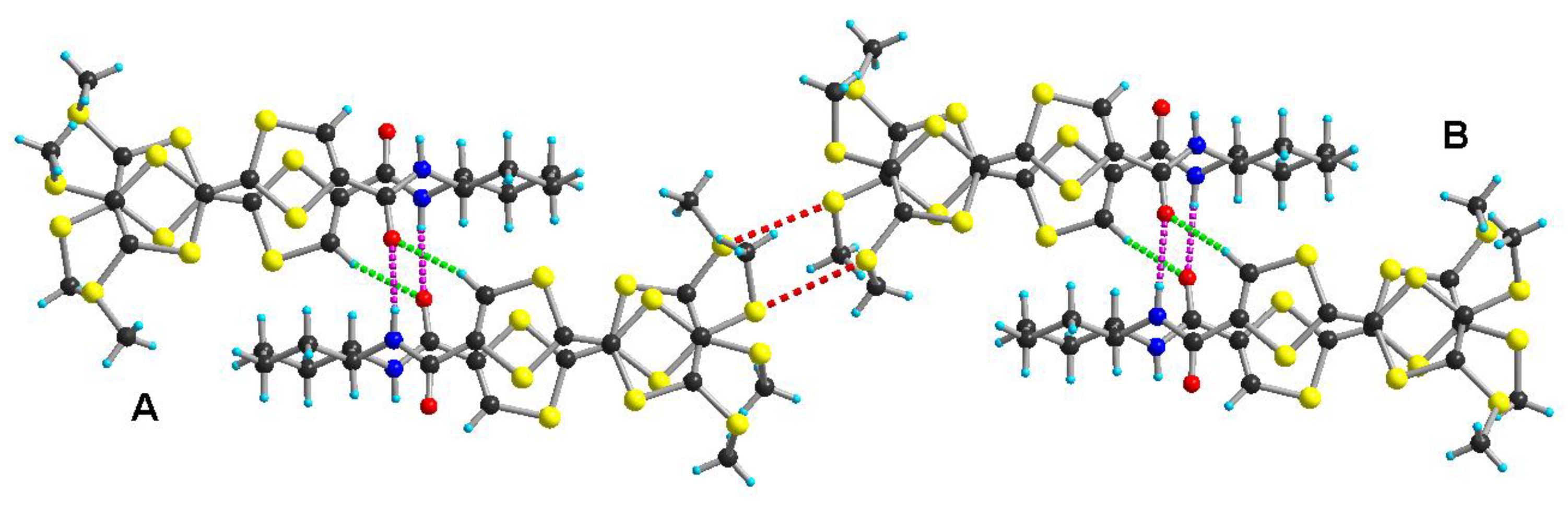
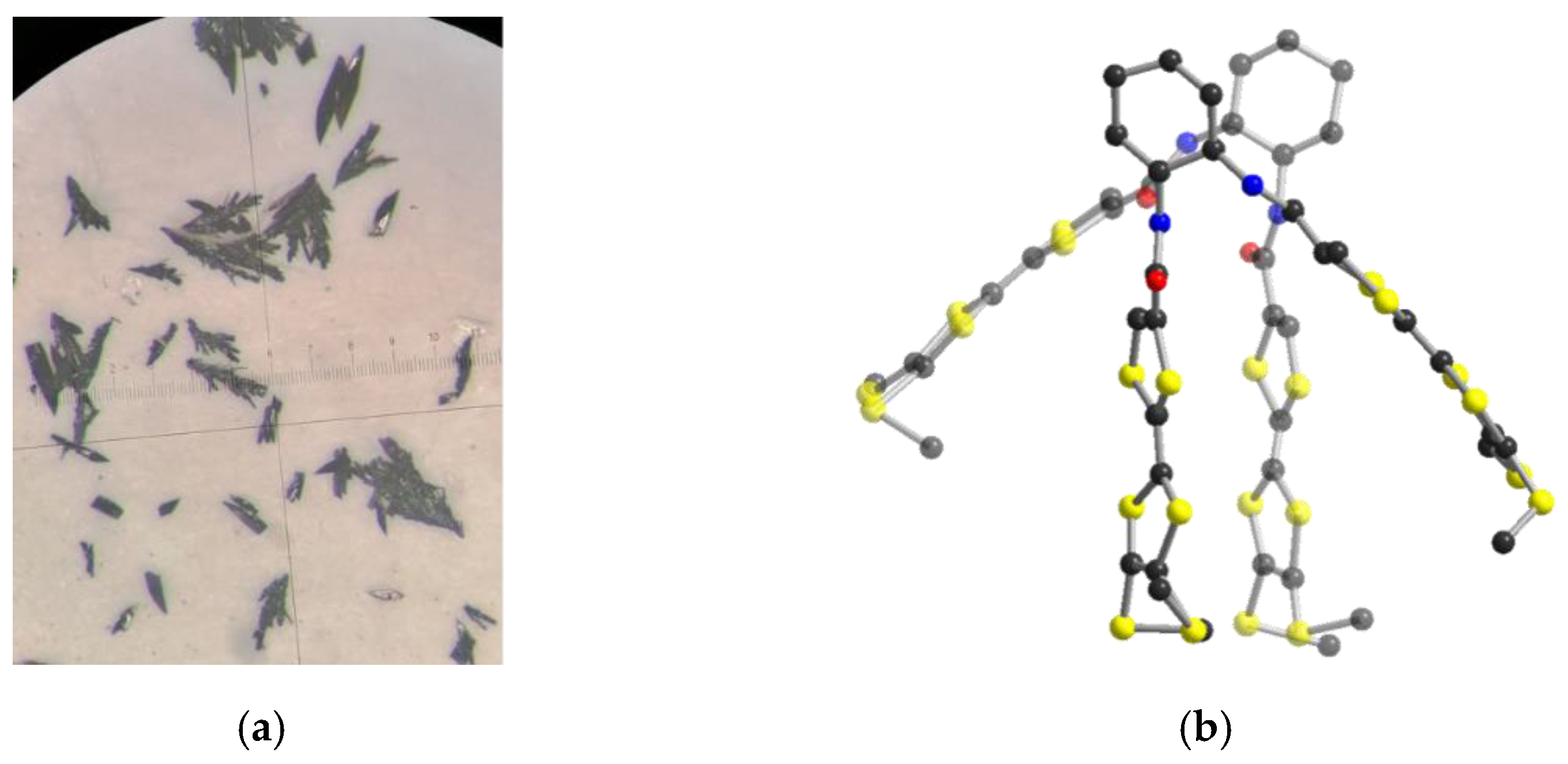
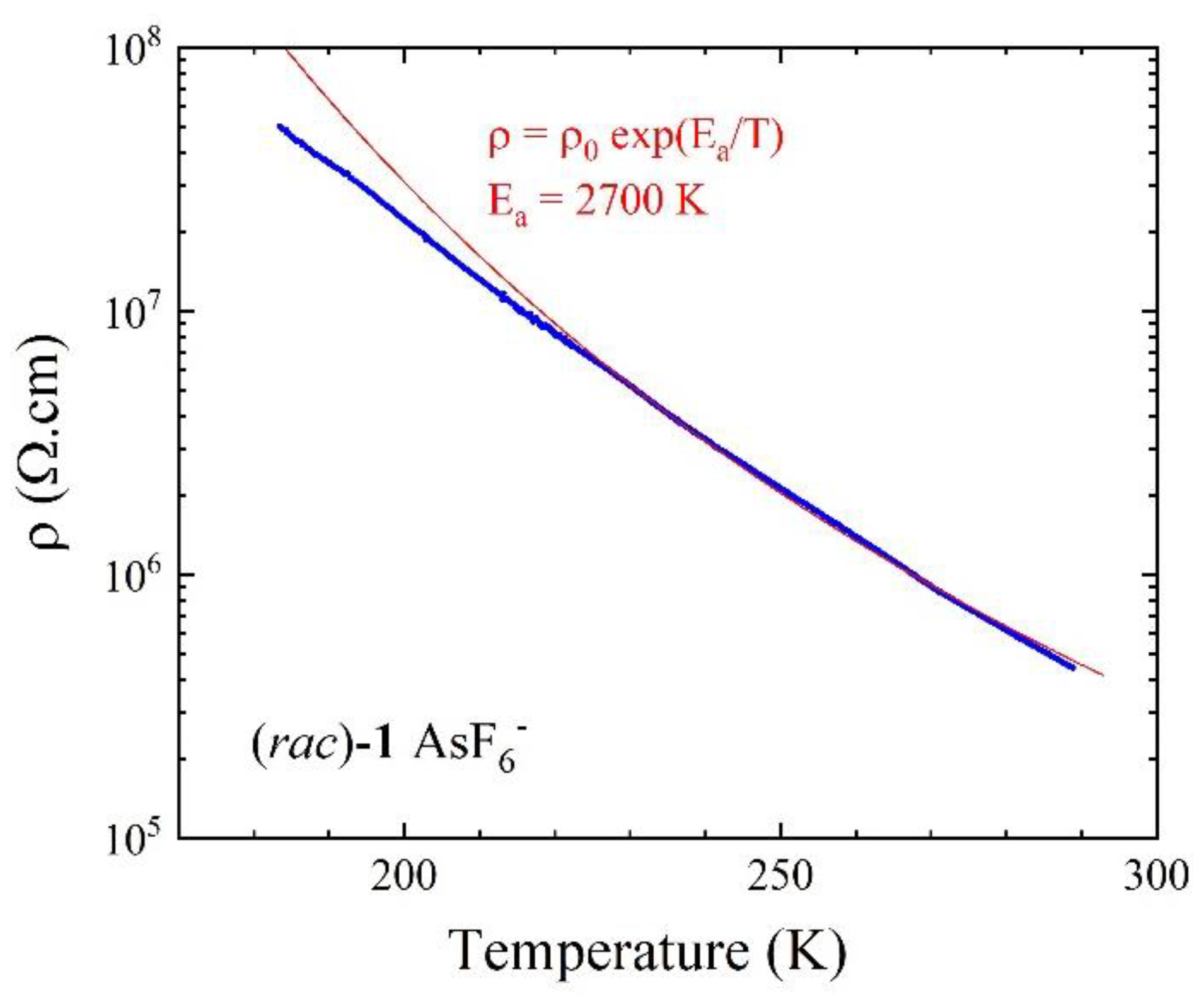
2.2. Electrocrystallization of Donor 1
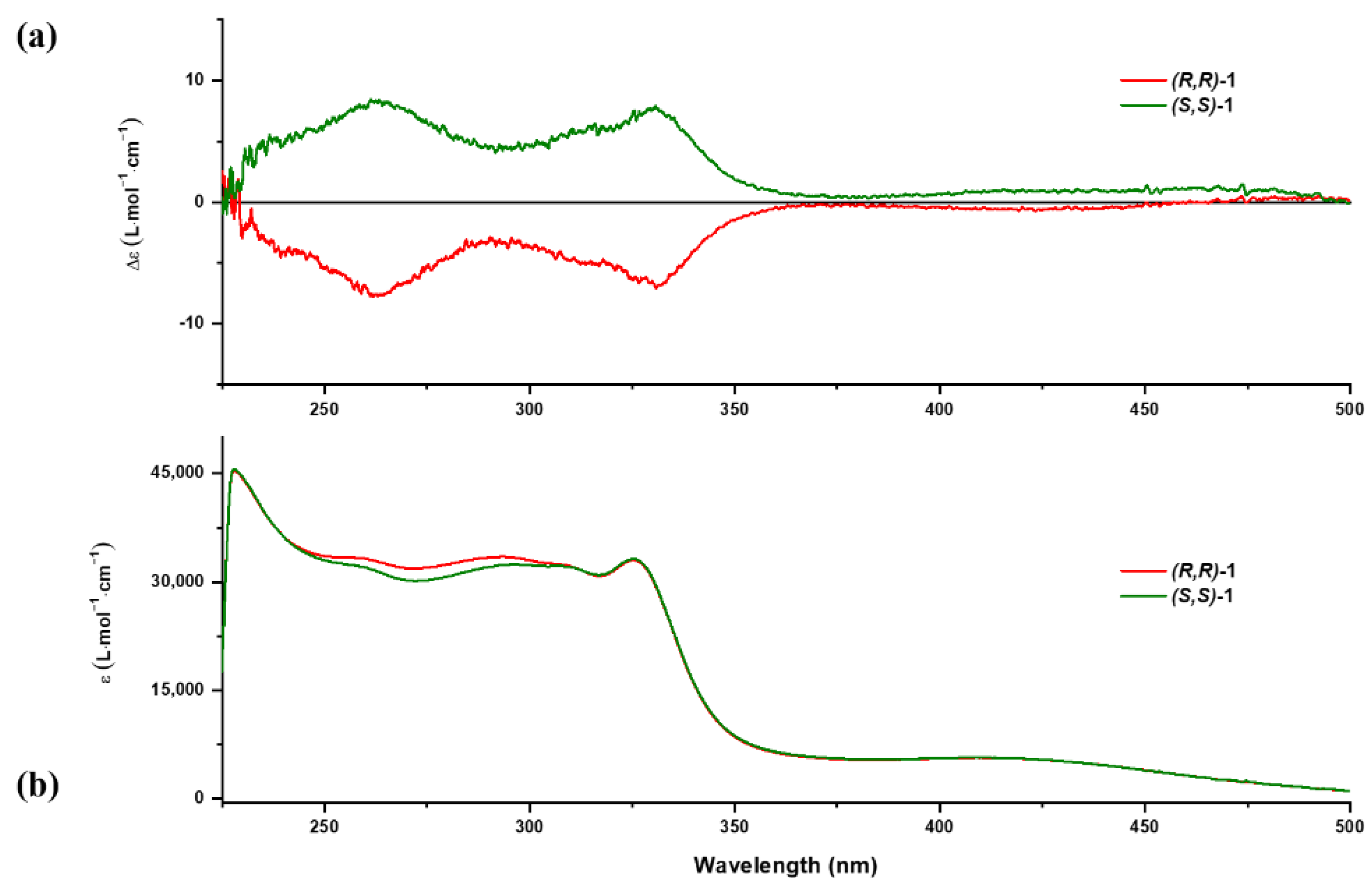
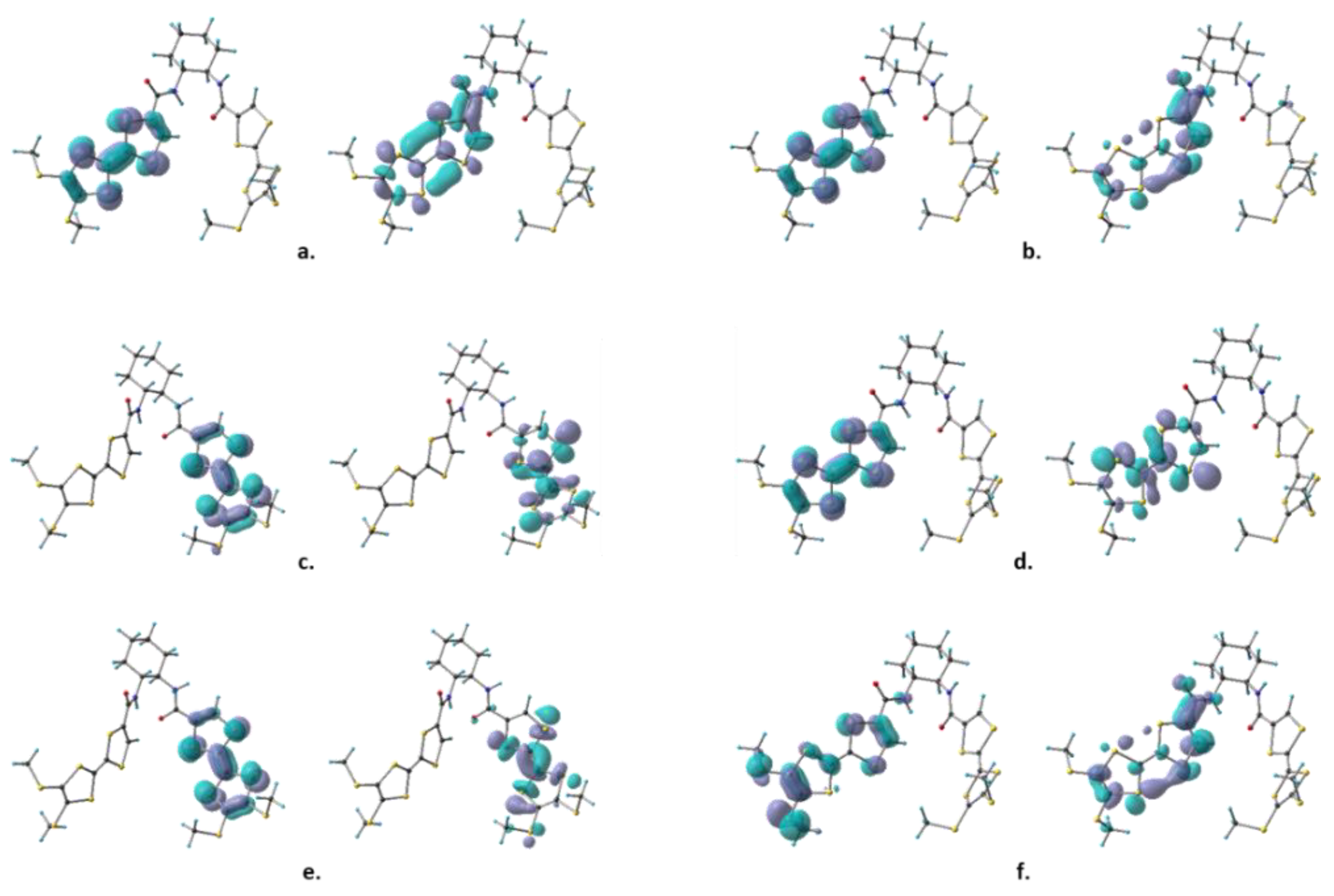
2.3. DFT and TD-DFT Calculations
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Avarvari, N.; Wallis, J.D. Strategies towards Chiral Molecular Conductors. J. Mater. Chem. 2009, 19, 4061–4076. [Google Scholar] [CrossRef] [Green Version]
- Wallis, J.D.; Karrer, A.; Dunitz, J.D. Chiral metals? A chiral substrate for organic conductors and superconductors. Helv. Chim. Acta 1986, 69, 69–70. [Google Scholar] [CrossRef]
- Karrer, A.; Wallis, J.D.; Dunitz, J.D.; Hilti, B.; Mayer, C.W.; Bürkle, M.; Pfeiffer, J. Structures and Electrical Properties of Some New Organic Conductors Derived from the Donor Molecule TMET (S,S,S,S-Bis(dimethylethylenedithio) tetrathiafulvalene). Helv. Chim. Acta 1987, 70, 942–953. [Google Scholar] [CrossRef]
- Rikken, G.L.J.A.; Fölling, J.; Wyder, P. Electrical Magnetochiral Anisotropy. Phys. Rev. Lett. 2001, 87, 236602. [Google Scholar] [CrossRef] [PubMed]
- Krstić, V.; Roth, S.; Burghard, M.; Kern, K.; Rikken, G.L.J.A. Magneto-Chiral Anisotropy in Charge Transport Through Single-Walled Carbon Nanotubes. J. Chem. Phys. 2002, 117, 11315–11319. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.J.A.; Avarvari, N. Electrical magneto-chiral anisotropy in a bulk chiral molecular conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, F.; Zigon, N.; Avarvari, N. Main-Group-Based Electro- and Photoactive Chiral Materials. Chem. Rev. 2019, 119, 8435–8478. [Google Scholar] [CrossRef] [PubMed]
- Heuzé, K.; Fourmigué, M.; Batail, P. The Crystal Chemistry of Amide-Functionalized Ethylenedithiotetrathiafulvalenes: EDT-TTF-CONRR’ (R, R’ = H, Me). J. Mater. Chem. 1999, 9, 2373–2379. [Google Scholar] [CrossRef]
- Heuzé, K.; Fourmigué, M.; Batail, P.; Canadell, E.; Auban-Senzier, P. Directing the Structures and Collective Electronic Properties of Organic Conductors: The Interplay of π-Overlap Interactions and Hydrogen Bonds. Chem. Eur. J. 1999, 5, 2971–2976. [Google Scholar] [CrossRef]
- Heuzé, K.; Mézière, C.; Fourmigué, M.; Batail, P.; Coulon, C.; Canadell, E.; Auban-Senzier, P.; Jérome, D. An Efficient, Redox-Enhanced Pair of Hydrogen-Bond Tweezers for Chloride Anion Recognition, a Key Synthon in the Construction of a Novel Type of Organic Metal based on the Secondary Amide-Functionalized Ethylenedithiotetrathiafulvalene, ”-(EDT-TTF-CONHMe)2[Cl·H2O]. Chem. Mater. 2000, 12, 1898–1904. [Google Scholar]
- Devic, T.; Avarvari, N.; Batail, P. A Series of Redox Active, Tetrathiafulvalene-Based Amido-Pyridines and Bipyridines Ligands: Syntheses, Crystal Structures, Radical Cation Salt and Group 10 Transition Metal Complexes. Chem. Eur. J. 2004, 10, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, R.; Wu, G.; Sugawa, K.; Otsuki, J. Intermolecular Electronic Communication in Tetrathiafulvalene Derivatives with Hydrogen-Bonding Amide Units. Asian J. Org. Chem. 2018, 7, 897–901. [Google Scholar] [CrossRef]
- Devic, T.; Rondeau, D.; Şahin, Y.; Levillain, E.; Clérac, R.; Batail, P.; Avarvari, N. Copper(I/II) complexes of a bis(tetrathiafulvalene)-2,2’-bipyridine: Synthesis, characterization, magnetic and electrochemical properties. Dalton Trans. 2006, 10, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Baudron, S.A.; Avarvari, N.; Batail, P.; Coulon, C.; Clérac, R.; Canadell, E.; Auban-Senzier, P. Singular Crystalline β’-Layered Topologies Directed by Ribbons of Self-Complementary Amide···Amide Ring Motifs in [EDT-TTF-(CONH2)2]2X (X = HSO4−, ClO4−, ReO4−, AsF6−): Coupled Activation of Ribbon Curvature, Electron Interactions, and Magnetic Susceptibility. J. Am. Chem. Soc. 2003, 125, 11583–11590. [Google Scholar] [PubMed]
- Baudron, S.A.; Avarvari, N.; Canadell, E.; Auban-Senzier, P.; Batail, P. Structural Isomerism in Crystals of Redox-Active Secondary ortho-Diamides: The Role of Competing Intra- and Intermolecular Hydrogen Bonds in Directing Crystalline Topologies. Chem. Eur. J. 2004, 10, 4498–4511. [Google Scholar] [CrossRef]
- Shi, Z.; Lu, Z.-J.; Zhu, Q.-Y.; Huo, L.-B.; Han, Q.-H.; Bian, G.-Q.; Dai, J. Diamino-Diamido Tetrathiafulvalene for the Sensing of Anions and Cations: A View in Electrochemistry and Structure. J. Phys. Chem. B 2011, 115, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Fourmigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef] [PubMed]
- Mroweh, N.; Pop, F.; Mézière, C.; Allain, M.; Auban-Senzier, P.; Vanthuyne, N.; Alemany, P.; Canadell, E.; Avarvari, N. Combining Chirality and Hydrogen Bonding in Methylated Ethylenedithio-Tetrathiafulvalene Primary Diamide Precursors and Radical Cation Salts. Cryst. Growth Des. 2020, 20, 2516–2526. [Google Scholar] [CrossRef]
- Awheda, I.; Krivickas, S.J.; Yang, S.; Martin, L.; Guziak, M.A.; Brooks, A.C.; Pelletier, F.; Le Kerneau, M.; Day, P.; Horton, P.N.; et al. Synthesis of new chiral organosulfur donors with hydrogen bonding functionality and their first charge transfer salts. Tetrahedron 2013, 69, 8738–8750. [Google Scholar] [CrossRef] [Green Version]
- Short, J.I.; Blundell, T.J.; Krivickas, S.J.; Yang, S.; Wallis, J.D.; Akutsu, H.; Nakazawa, Y.; Martin, L. Chiral molecular conductor with an insulator–metal transition close to room temperature. Chem. Commun. 2020, 56, 9497–9500. [Google Scholar] [CrossRef] [PubMed]
- Réthoré, C.; Fourmigué, M.; Avarvari, N. Chiral Tetrathiafulvalene-Hydroxyamides and -Oxazolines: Hydrogen Bonding, Chirality, and a Radical Cation Salt. Tetrahedron 2005, 61, 10935–10942. [Google Scholar] [CrossRef]
- Riobé, F.; Avarvari, N. C2-Symmetric chiral tetrathiafulvalene-bis(oxazolines) (TTF-BOX): New precursors for organic materials and electroactive metal complexes. Chem. Commun. 2009, 25, 3753–3755. [Google Scholar] [CrossRef]
- Réthoré, C.; Fourmigué, M.; Avarvari, N. Tetrathiafulvalene based phosphino-oxazolines: A new family of redox active chiral ligands. Chem. Commun. 2004, 12, 1384–1385. [Google Scholar] [CrossRef] [PubMed]
- Réthoré, C.; Avarvari, N.; Canadell, E.; Auban-Senzier, P.; Fourmigué, M. Chiral Molecular Metals: Syntheses, Structures and Properties of the AsF6− Salts of Racemic (+/−), (R)- and (S)-Tetrathiafulvalene-Oxazoline Derivatives. J. Am. Chem. Soc. 2005, 127, 5748–5749. [Google Scholar] [CrossRef]
- Saad, A.; Jeannin, O.; Fourmigué, M. Helical Organization of Chiral Binaphthyl Tetrathiafulvalene Primary Amides through Hydrogen Bonding Interactions. CrystEngComm 2010, 12, 3866–3874. [Google Scholar] [CrossRef] [Green Version]
- Tatewaki, Y.; Hatanaka, T.; Tsunashima, R.; Nakamura, T.; Kimura, M.; Shirai, H. Conductive Nanoscopic Fibrous Assemblies Containing Helical Tetrathiafulvalene Stacks. Chem. Asian J. 2009, 4, 1474–1479. [Google Scholar] [CrossRef]
- Danila, I.; Riobé, F.; Piron, F.; Puigmartí-Luis, J.; Wallis, J.D.; Linares, M.; Ågren, H.; Beljonne, D.; Amabilino, D.B.; Avarvari, N. Hierarchical Chiral Expression from the Nano- to Mesoscale in Synthetic Supramolecular Helical Fibers of a Nonamphiphilic C3-Symmetrical π-Functional Molecule. J. Am. Chem. Soc. 2011, 133, 8344–8353. [Google Scholar] [CrossRef]
- Danila, I.; Pop, F.; Escudero, C.; Feldborg, L.N.; Puigmartí-Luis, J.; Riobé, F.; Avarvari, N.; Amabilino, D.B. Twists and Turns in the Hierarchical Self-Assembly Pathways of a non-Amphiphilic Chiral Supramolecular Material. Chem. Commun. 2012, 48, 4552–4554. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Melan, C.; Danila, I.; Linares, M.; Beljonne, D.; Amabilino, D.B.; Avarvari, N. Hierarchical Self-Assembly of Supramolecular Helical Fibres from Amphiphilic C3-Symmetrical Functional Tris(tetrathiafulvalenes). Chem. Eur. J. 2014, 20, 17443–17453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5651. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Cauchy, T.; Pop, F.; Cuny, J.; Avarvari, N. Conformational Study and Chiroptical Properties of Chiral Dimethyl-Ethylenedithio-Tetrathiafulvalene (DM-EDT-TTF). Chimia 2018, 72, 389–393. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (rac)-1 | |
|---|---|
| Formula | C24H24N2O2S12 |
| M [g∙mol−1] | 757.17 |
| T [K] | 150.01(10) |
| Crystal system | triclinic |
| Space group | P–1 |
| a [Å] | 9.0042(7) |
| b [Å] | 16.8256(13) |
| c [Å] | 21.7297(10) |
| α [°] | 78.082(5) |
| β [°] | 85.629(5) |
| γ [°] | 80.197(6) |
| V [Å3] | 3171.2(4) |
| Z | 4 |
| ρcalcd [g cm−3] | 1.586 |
| μ [mm−1] | 7.917 |
| Goodness-of-fit on F2 | 1.183 |
| Final R1 a/wR2 b [I > 2σ(I)] | 0.1109/0.3032 |
| R1 a/wR2 b (all data) | 0.1708/0.4094 |
| CCDC number | 2206330 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdan, A.; Moraru, I.-T.; Auban-Senzier, P.; Grosu, I.; Pop, F.; Avarvari, N. Chiral Bis(tetrathiafulvalene)-1,2-cyclohexane-diamides. Molecules 2022, 27, 6926. https://doi.org/10.3390/molecules27206926
Bogdan A, Moraru I-T, Auban-Senzier P, Grosu I, Pop F, Avarvari N. Chiral Bis(tetrathiafulvalene)-1,2-cyclohexane-diamides. Molecules. 2022; 27(20):6926. https://doi.org/10.3390/molecules27206926
Chicago/Turabian StyleBogdan, Alexandra, Ionuț-Tudor Moraru, Pascale Auban-Senzier, Ion Grosu, Flavia Pop, and Narcis Avarvari. 2022. "Chiral Bis(tetrathiafulvalene)-1,2-cyclohexane-diamides" Molecules 27, no. 20: 6926. https://doi.org/10.3390/molecules27206926