
Metal Coordination Effects on the Photophysics of Dipyrrinato Photosensitizers



Abstract
:
1. Introduction
2. Photodynamic Therapy
2.1. Mechanism
2.2. Triplet Photosensitizers
3. The Heavy Atom Effect in BODIPYs and Chlorins
4. Transition Metal Triplet Photosensitizers
4.1. Characterization Techniques for Triplet Photosensitizers
4.1.1. Spectroscopic Studies
4.1.2. DFT and TD-DFT Calculations
4.2. Enhancing TM Complexes as Triplet Photosensitizers
4.2.1. Enhancing the Molar Absorption Coefficient
4.2.2. Enhancing Triplet Excited State Lifetime
4.2.3. Practical Example: Ru(II) Photosensitizer TLD-1433
5. Dipyrrinato Complexes as PSs for PDT
5.1. d-Block Dipyrrinato Metal Complexes
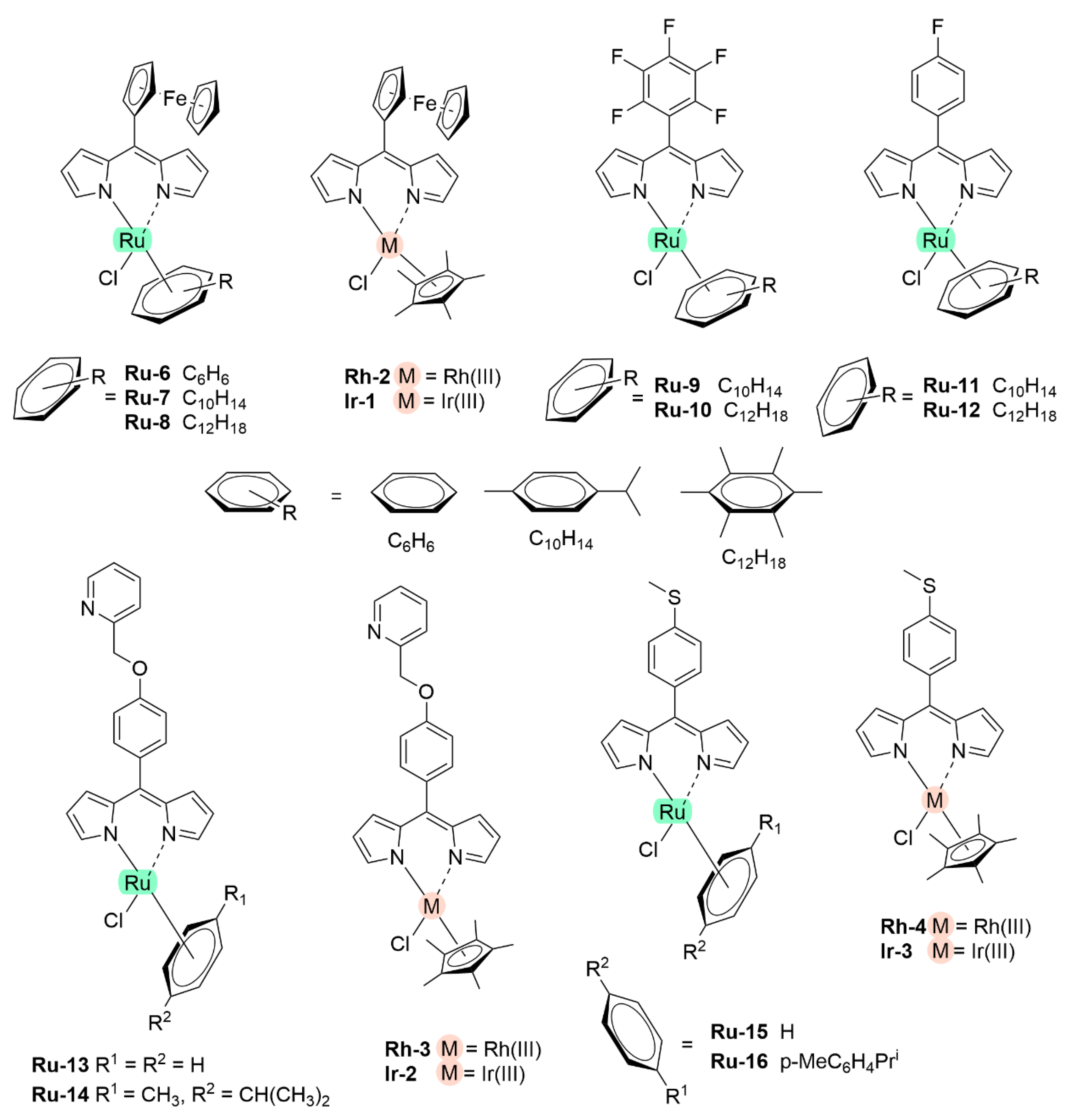
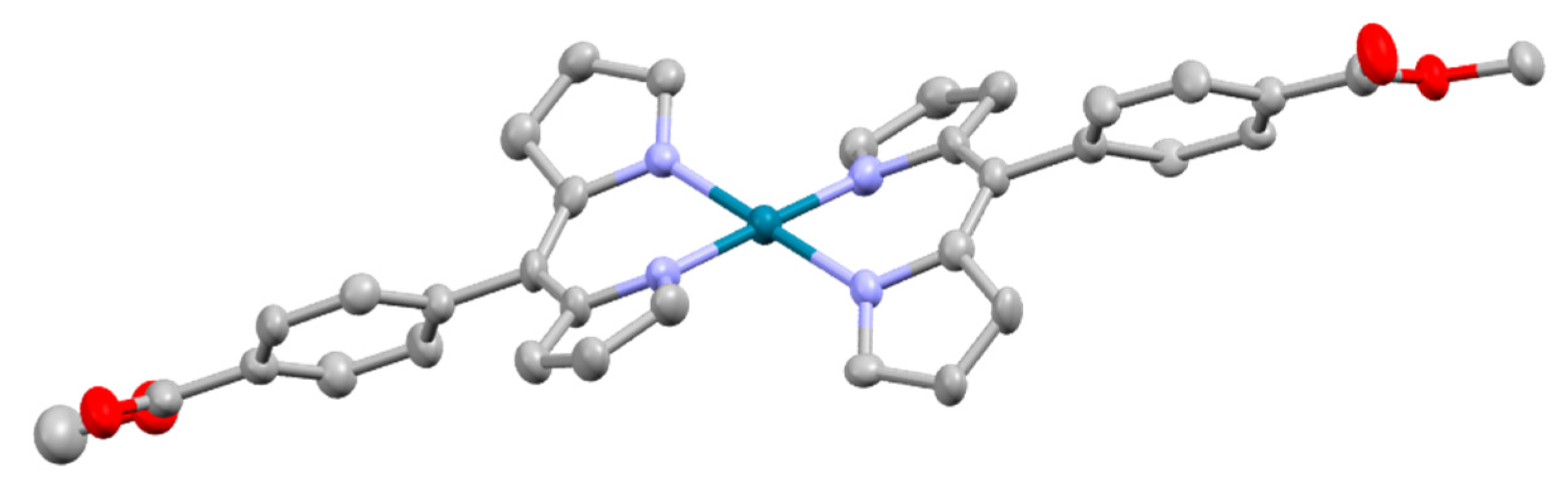
5.1.1. Re(I), Ru(II), Rh(III) and Ir(III) Dipyrrinato Complexes
5.1.2. Dipyrrinato-Zn(II) Complexes
5.1.3. (Dipyrrinato)-Pd(II) and -Pt(II) Complexes
5.1.4. Bis(dipyrrinato)Ni(II) and -Cu(II) Complexes
5.1.5. Further Comparisons between d-Block Metal Dipyrrinato Complexes
5.2. p-Block Dipyrrinato Complexes
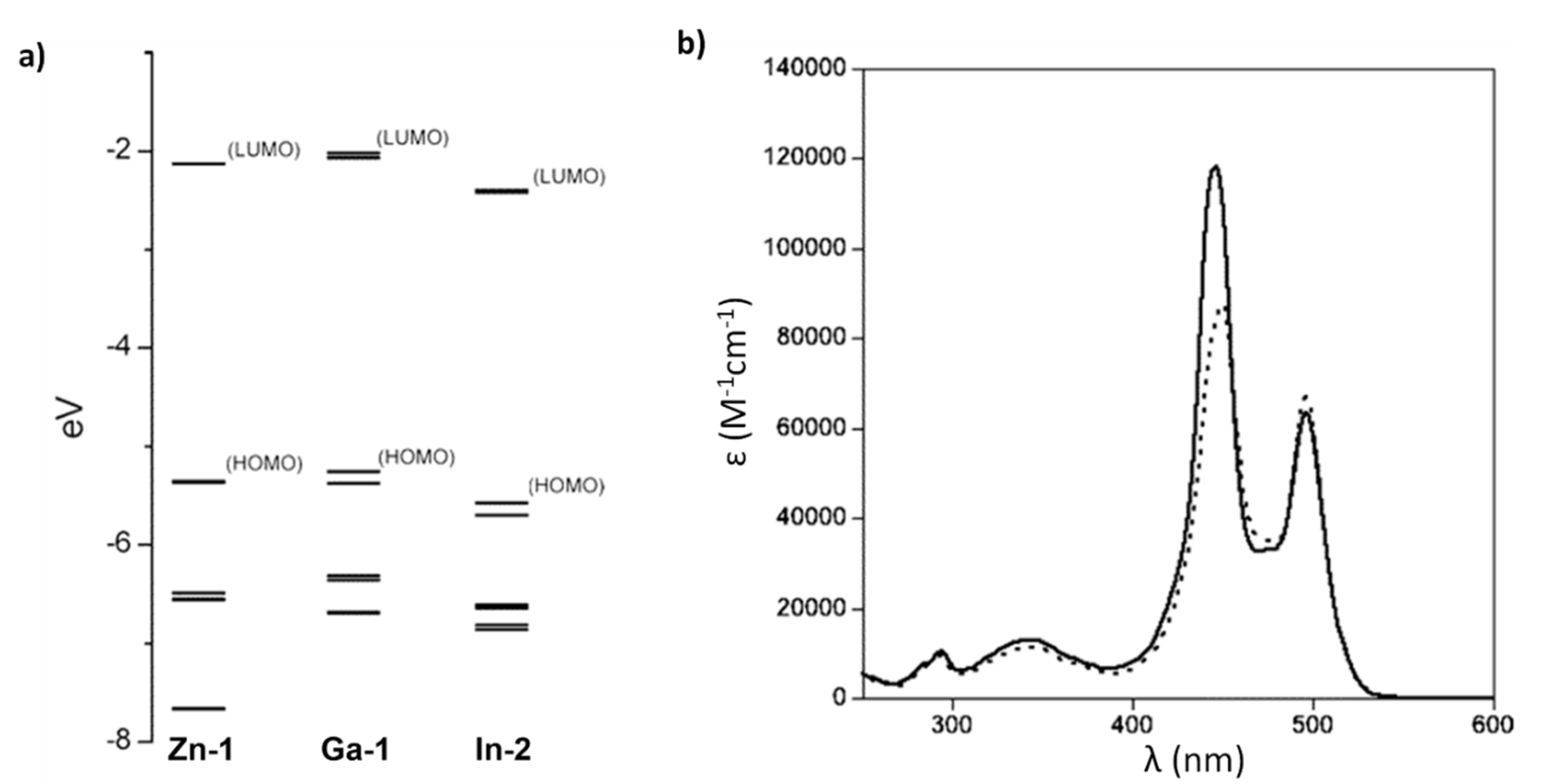
5.2.1. Dipyrrinato-Ga(III) and -In(III) Complexes
5.2.2. Dipyrrinato-Al(III) Complexes
6. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations and Symbols
| A | Absorbance at irradiation wavelength |
| Bipy | 2,2′-bipyridine |
| BODIPY | 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene |
| CT | Charge transfer |
| CycHex | Cyclohexane |
| DCM | Dichloromethane |
| DFT | Density functional theory |
| DIPY | Heteroleptic p-block mono(dipyrrinato) complex |
| DPM | Dipyrromethene |
| EnT | Energy transfer |
| F | Fraction of light absorbed |
| HSO | Spin–orbital Hamiltonian |
| HAE | Heavy atom effect |
| Hex | Mixture of hexanes |
| MCH | Methylcyclohexane |
| HOMO | Highest occupied molecular orbital |
| HOMO-1 | MO level one step lower in energy than the HOMO |
| I | Integrated emission intensity |
| I0 | Light intensity of the irradiation source |
| IC | Internal Conversion |
| 1,3IL | Singlet or triplet intraligand state |
| 1,3ILCT | Singlet or triplet intraligand charge transfer state |
| ISC | Intersystem crossing |
| Fluorescence rate constant | |
| Intersystem crossing rate constant | |
| Nonradiative rate constant; | |
| Phosphorescence rate constant | |
| Bimolecular quenching constant | |
| Radiative rate constant; | |
| Stern-Volmer constant | |
| 1,3LLCT | Singlet or triplet ligand-to-ligand charge transfer, also called ICT state |
| 1,3LMCT | Singlet or triplet ligand-to-metal charge transfer |
| LUMO | Lowest unoccupied molecular orbital |
| LUMO + 1 | MO level one step higher in energy than the LUMO |
| 1,3MC | Singlet or triplet metal-centered |
| MeOH | Methanol |
| Mes | Mesityl |
| 1,3MLCT | Singlet or triplet metal-to-ligand charge transfer state |
| 1,3MMCT | Singlet or triplet metal-to-metal charge transfer state |
| MO | Molecular orbital |
| MOF | Metal-organic framework |
| n | Refractive index |
| NIR | Near-infrared region |
| PDT | Photodynamic therapy |
| Ph | Phenyl |
| PS | Photosensitizer |
| Q | Quencher (= 3O2 in PDT) |
| QY | Quantum yield, see φ |
| r3 | Mean cubic radial distribution of the electron |
| ROS | Reactive oxygen species |
| SBCT | Symmetry breaking charge transfer |
| SOC | Spin–orbital coupling |
| S0 | Singlet ground state |
| S1 | First singlet excited state |
| TA | Transient absorption |
| TDDFT | Time dependent DFT |
| THF | Tetrahydrofuran |
| TM | Transition metal |
| Tol | Toluene |
| TTA | Triplet-triplet annihilation |
| T1 | First triplet excited state |
| UV-Vis | Ultra-violet – visible range |
| Z | Atomic number |
| ∆λ | Stokes shift in nm |
| ∆ES1−T1 | Energy gap between the first triplet excited state and the first singlet excited state |
| ∆ET1−S0 | Energy gap between the first triplet excited state and the ground state |
| ∆ṽ | Stokes shift in cm−1 |
| ∆ṽ0 | Uncorrected Stokes shift in cm−1, obtained from eq. Equation (6) |
| ɛ | Molar absorption coefficient |
| λ | Wavelength (in cm−1) |
| λa | Wavelength at maximum absorption |
| λex | Excitation wavelength |
| λf | Wavelength at maximum emission |
| τ | Lifetime of a state |
| τ0 | Lifetime in the absence of Q |
| τr | Radiative lifetime |
| τS | Singlet state lifetime |
| τT | Triplet state lifetime |
| Quantum yield (QY) | |
| Singlet oxygen quantum yield | |
| Fluorescence quantum yield | |
| Intersystem crossing quantum yield | |
| Quantum yield of triplet formation |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zn(II) Complex | Solvent | λa (nm) | ɛ a | λf (nm) | ∆ṽ0 b (∆ṽref) c (cm−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| Zn-2b | Tol | 485 | 1.15 | 501 | 658 (660) | 0.006 | [161,162] |
| Zn-2g | CycHex | 484 | 501 | 701(638) | 0.47 | [164] | |
| Tol | 487 | 1.15 | 500 | 534 (580) | 0.36 | [162] | |
| Tol | 486 | 1.15 | 503 | 695 (659) | 0.33 | [164] | |
| DCM | 485 | 495 | 417 (429) | 0.017 | [164] | ||
| Zn-2e | Tol | 1.15 | - (940) | 0.007 | [162] | ||
| Zn-2h | THF | 485 | 516 | 1239 | [163] | ||
| Zn-2i | THF | 486 | 518 | 1271 | [163] | ||
| Zn-3a | Tol | 485 | 1.44 | 500 | 619 | 0.054 | [120] |
| Zn-3g | CycHex | 493 | 506 | 521 (501) | 0.66 | [164] | |
| Tol | 495 | 1.6 | 509 | 556 (567) | 0.19/0.28 | [164,165] | |
| DCM | 493 | 508 | 599 (706) | 0.01 | [164,165] | ||
| Zn-3d | DCM | 494.5 | 1.2 | [184] | |||
| Zn-3c | DCM | 491 | 1.4 | [184] | |||
| Zn-4b (R2 = H) | DCM | 486 | 1.23 | 512 | 1045 | 0.007 | [167] |
| Tol | 493 | 505 | 482 | 0.002 | [170] | ||
| Zn-4f (R2 = H) | Tol | 494 | 510 | 635 | 0.243 | [170] | |
| Zn-4g (R2 = H) | CycHex | 489 | 507 | 726 (676) | 0.163 | [164] | |
| Tol | 491 | 1.63 | 509 | 720 (736) | 0.138 | [164] | |
| Tol | 490 | 1.63 | 510 | 759 (759) | 0.129 | [166] | |
| Tol | 493 | 510 | 676 | 0.249 | [170] | ||
| DCM | 488 | 509 | 845 (819) | 0.01 | [164] | ||
| THF | 488 | 1.67 | 507 | 768 | 0.026 | [166] | |
| Zn-4i (R2 = H) | DCM | 487 | 1.18 | 514 | 1079 | 0.015 | [167] |
| Zn-4j (R2 = H) | DCM | 491 | 1.28 | 518 | 1062 | 0.007 | [167] |
| Tol | 495 | 520 | 971 | 0.005 | [170] | ||
| Zn-4k (R2 = H) | DCM | 489 | 1.10 | 517 | 1108 | 0.004 | [167] |
| Zn-4g (R2 = C2H5) | CycHex | 506 | 533 | 1001 (1026) | 0.174 | [164] | |
| Tol | 508 | 1.4 | 534 | 958 (958) | 0.134 | [164] | |
| Tol | 508 | 1.4 | 532 | 888 (920) | 0.20 | [165] | |
| DCM | 506 | 528 | 823 (831) | 0.00/0.05 | [164,165] | ||
| Zn-4f (R2 = TA) | Tol | 553 | 1.8 | 579 | 812 | 0.72 | [165] |
| DCM | 0.27 | [165] | |||||
| Zn-4b (R2 = I) | Tol | 516 | 540 | 861 | 0.003 b | [79] | |
| Zn-4f (R2 = I) | Tol | 516 | 535 | 688 | 0.042 b | [79] | |
| Zn-4g (R2 = I) | Tol | 517 | 535 | 651 | 0.045 b | [79] | |
| Tol | 516 | 1.72 | 532 | 583 | 0.02 | [166] | |
| THF | 513 | 1.70 | 529 | 590 | 0.0014 | [166] | |
| Zn-4j (R2 = I) | Tol | 516 | 550 | 1198 | 0.011 d | [79] | |
| Zn-5 | Tol | 545 | [168] |
| Zn(II) Complex | Solvent | Ref. | ||||
|---|---|---|---|---|---|---|
| Zn-2b | Tol | 0.07 a | [161,162] | |||
| Zn-2g | CycHex | 0.11 | 0.12 b | 16,000 | [164] | |
| Tol | 0.13 | 0.13 | 50,000 | [162] | ||
| Tol | 0.11 | 0.22 b | [164] | |||
| DCM | 0.008 | [164] | ||||
| Zn-3g | CycHex | 0.14 | 0.07 b | [164] | ||
| Tol | 0.05 | 0.21 b | [164,165] | |||
| DCM | 0.00 | [164,165] | ||||
| Zn-4b (R2 = H) | DCM | 0.01 | 4.600 | 0.10 | [167] | |
| Zn-4g (R2 = H) | CycHex | 0.11 | 0.56 | [164] | ||
| Tol | 0.07 | 0.41 b | [164] | |||
| DCM | 0.00 | [164] | ||||
| Zn-4i (R2 = H) | DCM | 0.03 | 1400 | 0.08 | [167] | |
| Zn-4j (R2 = H) | DCM | 0.004 | 295,000 | 0.62 | [167] | |
| Zn-4k (R2 = H) | DCM | 0.004 | 146,000 | 0.28 | [167] | |
| Zn-4g (R2 = C2H5) | CycHex | 0.09 | 0.43 | [164] | ||
| Tol | 0.06 | 0.36 b | [164] | |||
| Zn-4b (R2 = I) | MeOH | 107 (207) c | 0.10/0.06//- d | [79] | ||
| Zn-4f (R2 = I) | MeOH | 221 (353) c | 0.54/0.43//0.43 d | [79] | ||
| Zn-4g (R2 = I) | MeOH | 182 (356) c | 0.61/0.52//0.57 d | [79] | ||
| Zn-4j (R2 = I) | MeOH | 354 (559) c | 0.43/0.45//0.38 d | [79] | ||
| Zn-5 | e | 630 | [168] |
| Pd(II) Complex | Solvent | λa (nm) | ɛ a | λf (nm) | ∆ṽref | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| Pd-5 | DMSO | 483 | [67] | ||||||
| Pd-6 | DMSO | 483 | [67] | ||||||
| Pd-1 | MeCN | 477 | 1.78 | 525, 561 | 2565 | 0.008 | 44.76 | 0.170 | [178] |
| Pd-2 | MeCN | 487 | 5.89 | 530, 576 | 2722 | 0.012 | 28.97 | 0.081 | [178] |
| Pd-3 | MeCN | 484 | 0.76 | 532, 581 | 2486 | 0.003 | 31.56 | 0.092 | [178] |
| Pd-4 | MeCN | 480 | 1.48 | 525, 570 | 2696 | 0.023 | 67.61 | 0.095 | [178] |
| M(II) Complex | Solvent | λa (nm) | ɛ (104 M−1 cm−1) |
|---|---|---|---|
| Ni-1b | DCM | 466.0 | 3.72 |
| Ni-1c | DCM | 480.0 | 3.89 |
| Ni-2b | DCM | 513.0 | 6.17 |
| Ni-2c | DCM | 515.0 | 5.01 |
| Cu-1b | DCM | 466.0 | 6.31 |
| Cu-1c | DCM | 471.1 | 6.61 |
| Cu-2b | DCM | 504.0 | 7.08 |
| Cu-2c | DCM | 510.0 | 5.25 |
| Co-2b | DCM | 497.5 | 14.1 |
| Co-2c | DCM | 500.5 | 11.2 |
| Zn-3c | DCM | 491.0 | 13.8 |
| Zn-3d | DCM | 494.5 | 12.0 |
| Complex | Solvent | λa (nm) | ɛ a | λf (nm) | ∆ṽ (cm−1) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| B-4 | Tol | 604 | 6.11 | 650 | 1172 d | 0.35 | 2.53 | [203] | |
| DCM | 0.18 | 1.52 | [203] | ||||||
| Zn-2g | Hex | 487 b | 11.5 b | 501 | 574 d | 0.36 b | 3.51 | ~10 | [3] |
| Ga-2 | Hex | 448, 496 | 8.7, 6.8 | 528 | 1220 e | 0.024 | 3.75 | 156 | [3] |
| MCH | 3.76 | [3] | |||||||
| In-3 | Hex | 444, 496 | 11.7, 6.01 | 522 | 1113 e | 0.074 | 1.93 | 26 | [3] |
| MCH | 1.82 | [3] | |||||||
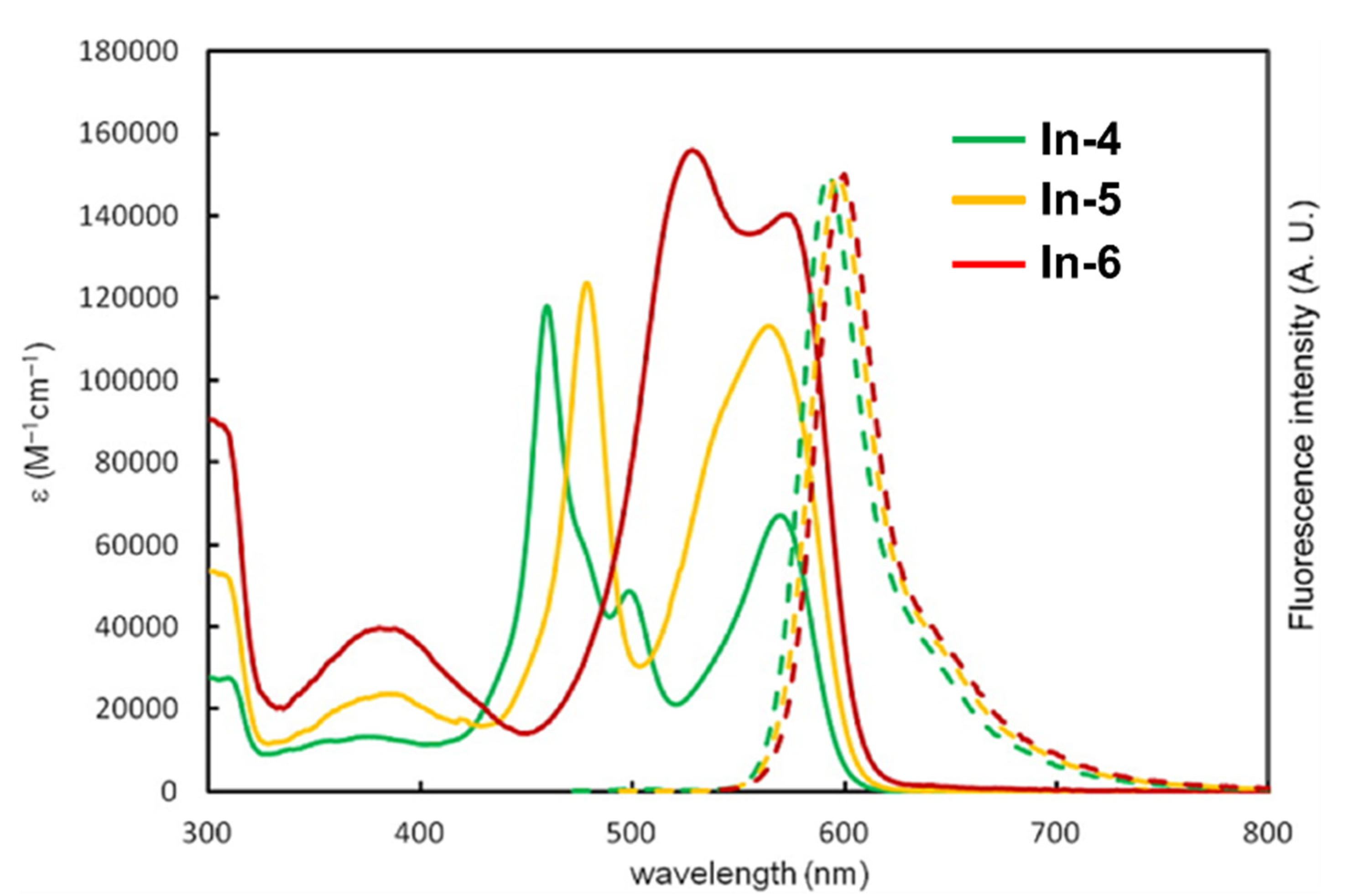
| In-4 | Tol | 459, 570 | 11.8, 6.73 | 592 | - | 0.41 | 2.44 | [203] | |
| DCM | 0.053 | 1.29 | [203] | ||||||
| In-5 | Tol | 478, 542 c, 564 | 12.4, 9.24 c, 11.3 | 596 | - | 0.34 | 2.53 | [203] | |
| DCM | 0.021 | 1.42 | [203] | ||||||
| In-6 | Tol | 528, 572 | 15.6, 14.0 | 600 | - | 0.28 | 2.49 | [203] | |
| DCM | 0.015 | 1.48 | [203] |
| DIPY | Solvent | λa (nm) | ɛ a | λf (nm) | ∆ṽ0 b [∆ṽref] c (cm−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| B-2 | MeOH | 502 | 12 | 508 | 235 | 0.70 | [55] |
| B-3 | MeOH | 534 | 11 | 548 | 478 | 0.02 | [55] |
| B-5 | Tol:MeOH (99:1) | 626 | 645 | 471 | 0.72 | [119] | |
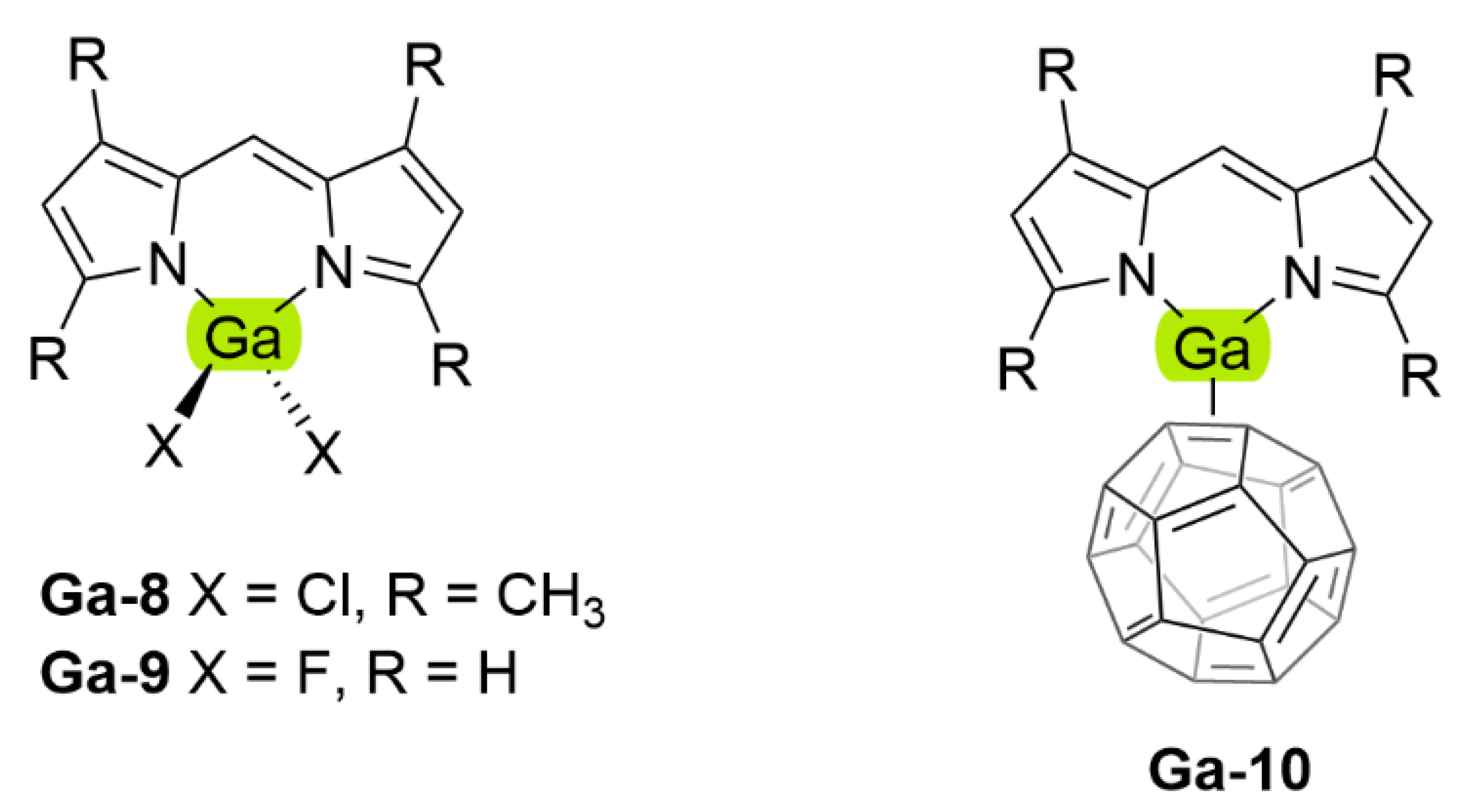
| Ga-8 | DCM | 494 | 501 | 283 | 0.82 | [113] | |
| Tol | 497 | 505 | 319 | 0.91 | [113] | ||
| Al-12 | Tol:MeOH (99:1) | 624 | 650 | 641 [560] | 0.23 | [119] | |
| Al-13 | Tol:MeOH (99:1) | 625 | 647 | 544 | 0.72 | [119] | |
| Al-14d | Tol:MeOH (99:1) | 607 | 620 | 345 [360] | 0.55 | [119] | |
| Al-15d | Tol:MeOH (99:1) | 607 | 620 | 345 | 0.56 | [119] | |
| Al-16d | Tol:MeOH (99:1) | 608 | 619 | 292 | 0.83 | [119] |
| 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| 2 | B | ||||||
| 3 | Al (III) d0 | ||||||
| 4 | Mn (II) d5 Mn (III) d4 Mn (VII)d0 | Fe (I) d7 Fe (II) d6 Fe (III) d5 | Co (I) d8 Co (II) d7 Co (III) d6 | Ni (II) d8 | Cu (I) d10 Cu (II) d9 Cu (III) d8 | Zn (II) d10 | Ga (III) d10 |
| 5 | Tc (I) d6 Tc (IV) d3 Tc (VII) d0 | Ru (II) d6 Ru (III) d5 Ru (IV) d4 | Rh (III) d6 | Pd (II) d8 | Ag (I) d10 Ag (II) d9 Ag (III) d8 | Cd (II) d10 | In (III) d10 |
| 6 | Re (I) d6 | Os (II) d6 Os (IV) d4 Os (VIII)d0 | Ir (III) d6 | Pt (II) d8 | Au (I) d10 Au (II) d9 Au (III) d8 | Hg (II) d10 | Tl (III) d10 |
References
- Williams, R.J.P. A Comparison of Types of Catalyst: The Quality of Metallo-Enzymes. J. Inorg. Biochem. 2008, 102, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Thoi, V.S.; Stork, J.R.; Magde, D.; Cohen, S.M. Luminescent Dipyrrinato Complexes of Trivalent Group 13 Metal Ions. Inorg. Chem. 2006, 45, 10688–10697. [Google Scholar] [CrossRef] [PubMed]
- Baudron, S.A. Dipyrrin Based Metal Complexes: Reactivity and Catalysis. Dalton Trans. 2020, 49, 6161–6175. [Google Scholar] [CrossRef]
- Wood, T.E.; Thompson, A. Advances in the Chemistry of Dipyrrins and Their Complexes. Chem. Rev. 2007, 107, 1831–1861. [Google Scholar] [CrossRef]
- Shikha Singh, R.; Prasad Paitandi, R.; Kumar Gupta, R.; Shankar Pandey, D. Recent Developments in Metal Dipyrrin Complexes: Design, Synthesis, and Applications. Coord. Chem. Rev. 2020, 414, 213269. [Google Scholar] [CrossRef]
- Baudron, S.A. Luminescent Dipyrrin Based Metal Complexes. Dalton Trans. 2013, 42, 7498–7509. [Google Scholar] [CrossRef]
- Sakamoto, R.; Iwashima, T.; Tsuchiya, M.; Toyoda, R.; Matsuoka, R.; Kögel, J.F.; Kusaka, S.; Hoshiko, K.; Yagi, T.; Nagayama, T.; et al. New Aspects in Bis and Tris(Dipyrrinato)Metal Complexes: Bright Luminescence, Self-Assembled Nanoarchitectures, and Materials Applications. J. Mater. Chem. A 2015, 3, 15357–15371. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Orth, H. Die Chemie des Pyrrols. II Band. Pyrrolfarbstoffe. 1 Hälfte, Porphyrine, Hämin, Bilirubin und ihre Abkömmlinge; Akademische Verlagsgesellschaft GMBH: Leipzig, Germany, 1937. [Google Scholar]
- Matsuoka, R.; Nabeshima, T. Functional Supramolecular Architectures of Dipyrrin Complexes. Front. Chem. 2018, 6, 349. [Google Scholar] [CrossRef] [Green Version]
- Treibs, A.; Kreuzer, F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Lu, H.; Shen, Z. Editorial: BODIPYs and Their Derivatives: The Past, Present and Future. Front. Chem. 2020, 8, 290. [Google Scholar] [CrossRef]
- Kamkaew, A.; Lim, S.H.; Lee, H.B.; Kiew, L.V.; Chung, L.Y.; Burgess, K. BODIPY Dyes in Photodynamic Therapy. Chem. Soc. Rev. 2012, 42, 77–88. [Google Scholar] [CrossRef]
- Kue, C.S.; Ng, S.Y.; Voon, S.H.; Kamkaew, A.; Chung, L.Y.; Kiew, L.V.; Lee, H.B. Recent Strategies to Improve Boron Dipyrromethene (BODIPY) for Photodynamic Cancer Therapy: An Updated Review. Photochem. Photobiol. Sci. 2018, 17, 1691–1708. [Google Scholar] [CrossRef]
- Karges, J.; Blacque, O.; Gasser, G. Metal Dipyrrin Complexes as Potential Photosensitizers for Photodynamic Therapy. Inorg. Chim. Acta 2020, 505, 119482. [Google Scholar] [CrossRef]
- McLean, T.M.; Moody, J.L.; Waterland, M.R.; Telfer, S.G. Luminescent Rhenium(I)-Dipyrrinato Complexes. Inorg. Chem. 2012, 51, 446–455. [Google Scholar] [CrossRef]
- Swavey, S.; Morford, K.; Tsao, M.; Comfort, K.; Kilroy, M.K. Heteroleptic Monometallic and Trimetallic Ruthenium(II) Complexes Incorporating a π-Extended Dipyrrin Ligand: Light-Activated Reactions with the A549 Lung Cancer Cell Line. J. Inorg. Biochem. 2017, 175, 101–109. [Google Scholar] [CrossRef]
- Hohlfeld, B.F.; Gitter, B.; Kingsbury, C.J.; Flanagan, K.J.; Steen, D.; Wieland, G.D.; Kulak, N.; Senge, M.O.; Wiehe, A. Dipyrrinato-Iridium(III) Complexes for Application in Photodynamic Therapy and Antimicrobial Photodynamic Inactivation. Chem. Eur. J. 2021, 27, 6440–6459. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in Photodynamic Therapy: Part Two-Cellular Signaling, Cell Metabolism and Modes of Cell Death. Photodiagn. Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Plaetzer, K.; Krammer, B.; Berlanda, J.; Berr, F.; Kiesslich, T. Photophysics and Photochemistry of Photodynamic Therapy: Fundamental Aspects. Lasers Med. Sci. 2009, 24, 259–268. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Senge, M.O.; Radomski, M.W. Platelets, Photosensitizers, and PDT. Photodiagn. Photodyn. Ther. 2013, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.R.; Hasan, T. Photodynamic Therapy: A New Antimicrobial Approach to Infectious Disease? Photochem. Photobiol. Sci. 2004, 3, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, M. Photodynamic Antimicrobial Chemotherapy (PACT). J. Antimicrob. Chemother. 1998, 42, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, W.; Sun, J.; Guo, S. Triplet Photosensitizers: From Molecular Design to Applications. Chem. Soc. Rev. 2013, 42, 5323–5351. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, J.; Xie, L.; Guo, H.; Li, Q. Thienyl-Substituted BODIPYs with Strong Visible Light-Absorption and Long-Lived Triplet Excited States as Organic Triplet Sensitizers for Triplet–Triplet Annihilation Upconversion. RSC Adv. 2012, 2, 3942–3953. [Google Scholar] [CrossRef]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Hatz, S.; Lambert, J.D.C.; Ogilby, P.R. Measuring the Lifetime of Singlet Oxygen in a Single Cell: Addressing the Issue of Cell Viability. Photochem. Photobiol. Sci. 2007, 6, 1106–1116. [Google Scholar] [CrossRef]
- Yao, Q.; Fan, J.; Long, S.; Zhao, X.; Li, H.; Du, J.; Shao, K.; Peng, X. The Concept and Examples of Type-III Photosensitizers for Cancer Photodynamic Therapy. Chem 2022, 8, 197–209. [Google Scholar] [CrossRef]
- Baskaran, R.; Lee, J.; Yang, S.-G. Clinical Development of Photodynamic Agents and Therapeutic Applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef]
- Nyman, E.S.; Hynninen, P.H. Research Advances in the Use of Tetrapyrrolic Photosensitizers for Photodynamic Therapy. J. Photochem. Photobiol. B Biol. 2004, 73, 1–28. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The Role of Porphyrin Chemistry in Tumor Imaging and Photodynamic Therapy. Chem. Soc. Rev. 2010, 40, 340–362. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef]
- Li, J.; Chen, T. Transition Metal Complexes as Photosensitizers for Integrated Cancer Theranostic Applications. Coord. Chem. Rev. 2020, 418, 213355. [Google Scholar] [CrossRef]
- Kaspler, P.; Mandel, A.; Dumoulin-White, R.; Roufaiel, M. Anticancer Photodynamic Therapy Using Ruthenium(II) and Os(II)-Based Complexes as Photosensitizers. In Tumor Progression and Metastasis; Lasfar, A., Cohen-Solal, K., Eds.; IntechOpen: Rijeka, Croatia, 2020; ISBN 978-1-78985-350-6. [Google Scholar]
- Karges, J. Combining Inorganic Chemistry and Biology: The Underestimated Potential of Metal Complexes in Medicine. ChemBioChem 2020, 21, 3044–3046. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Theralase Inc. A Phase II Clinical Study of Intravesical Photodynamic Therapy in Patients with BCG-Unresponsive Non-Muscle Invasive Bladder Cancer (“NMIBC”) or Patients Who Are Intolerant to BCG Therapy (“Study”); Theralase Inc.: Toronto, ON, Canada, 2022. Available online:https://clinicaltrials.gov/ (accessed on 10 October 2022).
- Mallidi, S.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Ichikawa, M.; Hasan, T. Beyond the Barriers of Light Penetration: Strategies, Perspectives and Possibilities for Photodynamic Therapy. Theranostics 2016, 6, 2458–2487. [Google Scholar] [CrossRef] [Green Version]
- Ben-Dror, S.; Bronshtein, I.; Wiehe, A.; Röder, B.; Senge, M.O.; Ehrenberg, B. On the Correlation Between Hydrophobicity, Liposome Binding and Cellular Uptake of Porphyrin Sensitizers. Photochem. Photobiol. 2006, 82, 695–701. [Google Scholar] [CrossRef]
- Calixto, G.M.F.; Bernegossi, J.; De Freitas, L.M.; Fontana, C.R.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for Photodynamic Therapy of Cancer: A Review. Molecules 2016, 21, 342. [Google Scholar] [CrossRef]
- Paszko, E.; Ehrhardt, C.; Senge, M.O.; Kelleher, D.P.; Reynolds, J.V. Nanodrug Applications in Photodynamic Therapy. Photodiagn. Photodyn. Ther. 2011, 8, 14–29. [Google Scholar] [CrossRef]
- Khurana, B.; Gierlich, P.; Meindl, A.; Gomes-da-Silva, L.C.; Senge, M.O. Hydrogels: Soft Matters in Photomedicine. Photochem. Photobiol. Sci. 2019, 18, 2613–2656. [Google Scholar] [CrossRef]
- Lehmann, P. Nebenwirkungen der topischen photodynamischen Therapie. Hautarzt 2007, 58, 597–603. [Google Scholar] [CrossRef]
- Callaghan, S.; Senge, M.O. The Good, the Bad, and the Ugly–Controlling Singlet Oxygen through Design of Photosensitizers and Delivery Systems for Photodynamic Therapy. Photochem. Photobiol. Sci. 2018, 17, 1490–1514. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Yao, H.-W.; Fu, Y.-J.; Guo, X.-F.; Wang, H. Effect of Substituents on Stokes Shift of BODIPY and Its Application in Designing Bioimaging Probes. Anal. Chim. Acta 2019, 1048, 194–203. [Google Scholar] [CrossRef]
- Balsukuri, N.; Lone, M.Y.; Jha, P.C.; Mori, S.; Gupta, I. Synthesis, Structure, and Optical Studies of Donor–Acceptor-Type Near-Infrared (NIR) Aza–Boron-Dipyrromethene (BODIPY) Dyes. Chem. Asian J. 2016, 11, 1572–1587. [Google Scholar] [CrossRef] [PubMed]
- Balsukuri, N.; Boruah, N.J.; Kesavan, P.E.; Gupta, I. Near Infra-Red Dyes Based on Pyrene Aza-BODIPYs. New J. Chem. 2018, 42, 5875–5888. [Google Scholar] [CrossRef]
- Marian, C.M. Spin–Orbit Coupling and Intersystem Crossing in Molecules. WIREs Comp. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Triplet State. Its Radiative and Nonradiative Properties. Acc. Chem. Res. 1968, 1, 8–16. [Google Scholar] [CrossRef]
- Pokhilko, P.; Krylov, A.I. Quantitative El-Sayed Rules for Many-Body Wave Functions from Spinless Transition Density Matrices. J. Phys. Chem. Lett. 2019, 10, 4857–4862. [Google Scholar] [CrossRef]
- Englman, R.; Jortner, J. The Energy Gap Law for Radiationless Transitions in Large Molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
- Braslavsky, S.E. Glossary of Terms Used in Photochemistry, 3rd Edition (IUPAC Recommendations 2006). Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Yogo, T.; Urano, Y.; Ishitsuka, Y.; Maniwa, F.; Nagano, T. Highly Efficient and Photostable Photosensitizer Based on BODIPY Chromophore. J. Am. Chem. Soc. 2005, 127, 12162–12163. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.J.; Agarrabeitia, A.R.; Duran-Sampedro, G.; Bañuelos Prieto, J.; Lopez, T.A.; Massad, W.A.; Montejano, H.A.; García, N.A.; Lopez Arbeloa, I. Synthesis and Functionalization of New Polyhalogenated BODIPY Dyes. Study of Their Photophysical Properties and Singlet Oxygen Generation. Tetrahedron 2012, 68, 1153–1162. [Google Scholar] [CrossRef]
- Laine, M.; Barbosa, N.A.; Kochel, A.; Osiecka, B.; Szewczyk, G.; Sarna, T.; Ziółkowski, P.; Wieczorek, R.; Filarowski, A. Synthesis, Structural, Spectroscopic, Computational and Cytotoxic Studies of BODIPY Dyes. Sensors Act. B Chem. 2017, 238, 548–555. [Google Scholar] [CrossRef]
- Lim, S.H.; Thivierge, C.; Nowak-Sliwinska, P.; Han, J.; van den Bergh, H.; Wagnières, G.; Burgess, K.; Lee, H.B. In Vitro and In Vivo Photocytotoxicity of Boron Dipyrromethene Derivatives for Photodynamic Therapy. J. Med. Chem. 2010, 53, 2865–2874. [Google Scholar] [CrossRef]
- Wong, W.-Y. Luminescent Organometallic Poly(Aryleneethynylene)s: Functional Properties towards Implications in Molecular Optoelectronics. Dalton Trans. 2007, 2007, 4495–4510. [Google Scholar] [CrossRef]
- Wong, W.-Y.; Ho, C.-L. Di-, Oligo- and Polymetallaynes: Syntheses, Photophysics, Structures and Applications. Coord. Chem. Rev. 2006, 250, 2627–2690. [Google Scholar] [CrossRef]
- Zhou, L.; Ge, X.; Liu, J.; Zhou, J.; Wei, S.; Li, F.; Shen, J. Internal Heavy Atom Effect of Au(III) and Pt(IV) on Hypocrellin A for Enhanced in Vitro Photodynamic Therapy of Cancer. Bioorg. Med. Chem. Lett. 2013, 23, 5317–5324. [Google Scholar] [CrossRef]
- Obata, M.; Hirohara, S.; Tanaka, R.; Kinoshita, I.; Ohkubo, K.; Fukuzumi, S.; Tanihara, M.; Yano, S. In Vitro Heavy-Atom Effect of Palladium(II) and Platinum(II) Complexes of Pyrrolidine-Fused Chlorin in Photodynamic Therapy. J. Med. Chem. 2009, 52, 2747–2753. [Google Scholar] [CrossRef]
- Melissari, Z.; Sample, H.C.; Twamley, B.; Williams, R.M.; Senge, M.O. Synthesis and Spectral Properties of Gem-Dimethyl Chlorin Photosensitizers. ChemPhotoChem 2020, 4, 601–611. [Google Scholar] [CrossRef]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-Based Photosensitizers for Photodynamic Therapy: The Future of Multimodal Oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef]
- Heinemann, F.; Karges, J.; Gasser, G. Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res. 2017, 50, 2727–2736. [Google Scholar] [CrossRef]
- Pucelik, B.; Sułek, A.; Dąbrowski, J.M. Bacteriochlorins and Their Metal Complexes as NIR-Absorbing Photosensitizers: Properties, Mechanisms, and Applications. Coord. Chem. Rev. 2020, 416, 213340. [Google Scholar] [CrossRef]
- Hall, J.D.; McLean, T.M.; Smalley, S.J.; Waterland, M.R.; Telfer, S.G. Chromophoric Dipyrrin Complexes Capable of Binding to TiO2: Synthesis, Structure and Spectroscopy. Dalton Trans. 2009, 39, 437–445. [Google Scholar] [CrossRef]
- Bosnich, B. Application of Exciton Theory to the Determination of the Absolute Configurations of Inorganic Complexes. Acc. Chem. Res. 1969, 2, 266–273. [Google Scholar] [CrossRef]
- Telfer, S.G.; McLean, T.M.; Waterland, M.R. Exciton Coupling in Coordination Compounds. Dalton Trans. 2011, 40, 3097–3108. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; El-Bayoumi, M.A. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Mooney, J.; Kambhampati, P. Get the Basics Right: Jacobian Conversion of Wavelength and Energy Scales for Quantitative Analysis of Emission Spectra. J. Phys. Chem. Lett. 2013, 4, 3316–3318. [Google Scholar] [CrossRef]
- Cormier, J.-F.; Fortin, M.; Frechette, J.; Noiseux, I.; Vernon, M.L.; Long, W. The Effects of Self-Absorption and Detection Geometry on Fluorescence Intensity and Decay Lifetime. Proc. SPIE 2005, 5702, 123–134. [Google Scholar]
- Porter, G.-N.; Norrish, R.G.W. Flash Photolysis and Spectroscopy. A New Method for the Study of Free Radical Reactions. Proc. R. Soc. London Ser. A Math. Phys. Sci. 1950, 200, 284–300. [Google Scholar] [CrossRef]
- Stern, O.; Volmer, M. Über Die Abklingungszeit Der Fluoreszenz. Physik. Z. 1919, 20, 183–188. [Google Scholar]
- Wilson, D.F. Oxygen Dependent Quenching of Phosphorescence: A Perspective. In Oxygen Transport to Tissue XIV; Erdmann, W., Bruley, D.F., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1992; pp. 195–201. ISBN 978-1-4615-3428-0. [Google Scholar]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Rate Constants for the Decay and Reactions of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. An Expanded and Revised Compilation. J. Phys. Chem. Ref. Data 1995, 24, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Garcìa-Fresnadillo, D.; Georgiadou, Y.; Orellana, G.; Braun, A.M.; Oliveros, E. Singlet-Oxygen (1Δg) Production by Ruthenium(II) Complexes Containing Polyazaheterocyclic Ligands in Methanol and in Water. Helv. Chim. Acta 1996, 79, 1222–1238. [Google Scholar] [CrossRef]
- Karges, J.; Basu, U.; Blacque, O.; Chao, H.; Gasser, G. Polymeric Encapsulation of Novel Homoleptic Bis(Dipyrrinato) Zinc(II) Complexes with Long Lifetimes for Applications as Photodynamic Therapy Photosensitisers. Angew. Chem. Int. Ed. 2019, 58, 14334–14340. [Google Scholar] [CrossRef] [PubMed]
- Crippa, P.R.; Vecli, A.; Viappiani, C. Time-Resolved Photoacoustic Spectroscopy: New Developments of an Old Idea: New Trends in Photobiology. J. Photochem. Photobiol. B Biol. 1994, 24, 3–15. [Google Scholar] [CrossRef]
- Heihoff, K.; Braslavsky, S.E. Triplet Lifetime Determination by Laser-Induced Optoacoustic Spectroscopy. Benzophenone/Iodide Revisited. Chem. Phys. Lett. 1986, 131, 183–188. [Google Scholar] [CrossRef]
- Suzen, S.; Gurer-Orhan, H.; Saso, L. Detection of Reactive Oxygen and Nitrogen Species by Electron Paramagnetic Resonance (EPR) Technique. Molecules 2017, 22, 181. [Google Scholar] [CrossRef]
- Gutsche, C.S.; Gräfe, S.; Gitter, B.; Flanagan, K.J.; Senge, M.O.; Kulak, N.; Wiehe, A. Pre-/Post-Functionalization in Dipyrrin Metal Complexes—Antitumor and Antibacterial Activity of Their Glycosylated Derivatives. Dalton Trans. 2018, 47, 12373–12384. [Google Scholar] [CrossRef]
- Vaz, G.M.F.; Paszko, E.; Davies, A.M.; Senge, M.O. High Content Screening as High Quality Assay for Biological Evaluation of Photosensitizers In Vitro. PLoS ONE 2013, 8, e70653. [Google Scholar] [CrossRef] [Green Version]
- Simone, B.C.D.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Metal Atom Effect on the Photophysical Properties of Mg(II), Zn(II), Cd(II), and Pd(II) Tetraphenylporphyrin Complexes Proposed as Possible Drugs in Photodynamic Therapy. Molecules 2017, 22, 1093. [Google Scholar] [CrossRef] [Green Version]
- Balabanov, N.B.; Peterson, K.A. Systematically Convergent Basis Sets for Transition Metals. I. All-Electron Correlation Consistent Basis Sets for the 3d Elements Sc–Zn. J. Chem. Phys. 2005, 123, 064107. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Basis Set Limit Electronic Excitation Energies, Ionization Potentials, and Electron Affinities for the 3d Transition Metal Atoms: Coupled Cluster and Multireference Methods. J. Chem. Phys. 2006, 125, 074110. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Puzzarini, C. Systematically Convergent Basis Sets for Transition Metals. II. Pseudopotential-Based Correlation Consistent Basis Sets for the Group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) Elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Dolg, M.; Stoll, H. Energy-Consistent Relativistic Pseudopotentials and Correlation Consistent Basis Sets for the 4d Elements Y–Pd. J. Chem. Phys. 2007, 126, 124101. [Google Scholar] [CrossRef]
- Figgen, D.; Peterson, K.A.; Dolg, M.; Stoll, H. Energy-Consistent Pseudopotentials and Correlation Consistent Basis Sets for the 5d Elements Hf–Pt. J. Chem. Phys. 2009, 130, 164108. [Google Scholar] [CrossRef]
- Dolg, M. Improved Relativistic Energy-Consistent Pseudopotentials for 3d-Transition Metals. Theor. Chem. Acc. 2005, 114, 297–304. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Tyson, D.S.; Castellano, F.N. Light-Harvesting Arrays with Coumarin Donors and MLCT Acceptors. Inorg. Chem. 1999, 38, 4382–4383. [Google Scholar] [CrossRef]
- Galletta, M.; Campagna, S.; Quesada, M.; Ulrich, G.; Ziessel, R. The Elusive Phosphorescence of Pyrromethene–BF2 Dyes Revealed in New Multicomponent Species Containing Ru(II)–Terpyridine Subunits. Chem. Commun. 2005, 2005, 4222–4224. [Google Scholar] [CrossRef]
- Cui, X.; Zhao, J.; Mohmood, Z.; Zhang, C. Accessing the Long-Lived Triplet Excited States in Transition-Metal Complexes: Molecular Design Rationales and Applications. Chem. Rec. 2016, 16, 173–188. [Google Scholar] [CrossRef]
- Bonnet, S. Shifting the Light Activation of Metallodrugs to the Red and Near-Infrared Region in Anticancer Phototherapy. Comments Inorg. Chem. 2015, 35, 179–213. [Google Scholar] [CrossRef]
- Reeßing, F.; Szymanski, W. Beyond Photodynamic Therapy: Light-Activated Cancer Chemotherapy. Curr. Med. Chem. 2017, 24, 4905–4950. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Rees, T.W.; Ke, L.; Ji, L.; Chao, H. Harnessing Ruthenium(II) as Photodynamic Agents: Encouraging Advances in Cancer Therapy. Coord. Chem. Rev. 2018, 363, 17–28. [Google Scholar] [CrossRef]
- Poynton, F.E.; Bright, S.A.; Blasco, S.; Williams, D.C.; Kelly, J.M.; Gunnlaugsson, T. The Development of Ruthenium(II) Polypyridyl Complexes and Conjugates for in Vitro Cellular and in Vivo Applications. Chem. Soc. Rev. 2017, 46, 7706–7756. [Google Scholar] [CrossRef]
- Knoll, J.D.; Albani, B.A.; Turro, C. Excited State Investigation of a New Ru(II) Complex for Dual Reactivity with Low Energy Light. Chem. Commun. 2015, 51, 8777–8780. [Google Scholar] [CrossRef] [Green Version]
- White, J.K.; Schmehl, R.H.; Turro, C. An Overview Of Photosubstitution Reactions Of Ru(II) Imine Complexes and Their Application in Photobiology and Photodynamic Therapy. Inorg. Chim. Acta 2017, 454, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.A.; Sadler, P.J. Photoactivatable Metal Complexes: From Theory to Applications in Biotechnology and Medicine. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2013, 371, 20120519. [Google Scholar] [CrossRef]
- Vos, J.G.; Kelly, J.M. Ruthenium Polypyridyl Chemistry; from Basic Research to Applications and Back Again. Dalton Trans. 2006, 2016, 4869–4883. [Google Scholar] [CrossRef]
- Zamora, A.; Vigueras, G.; Rodríguez, V.; Santana, M.D.; Ruiz, J. Cyclometalated Iridium(III) Luminescent Complexes in Therapy and Phototherapy. Coord. Chem. Rev. 2018, 360, 34–76. [Google Scholar] [CrossRef]
- Jiang, X.; Zhu, N.; Zhao, D.; Ma, Y. New Cyclometalated Transition-Metal Based Photosensitizers for Singlet Oxygen Generation and Photodynamic Therapy. Sci. China Chem. 2016, 59, 40–52. [Google Scholar] [CrossRef]
- Lazic, S.; Kaspler, P.; Shi, G.; Monro, S.; Sainuddin, T.; Forward, S.; Kasimova, K.; Hennigar, R.; Mandel, A.; McFarland, S.; et al. Novel Osmium-Based Coordination Complexes as Photosensitizers for Panchromatic Photodynamic Therapy. Photochem. Photobiol. 2017, 93, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Roundhill, D.M. Photochemistry, Photophysics, and Photoredox Reactions of Ru(Bpy)32+and Related Complexes. In Photochemistry and Photophysics of Metal Complexes; Roundhill, D.M., Ed.; Modern Inorganic Chemistry; Springer: Boston, MA, USA, 1994; pp. 165–215. ISBN 978-1-4899-1495-8. [Google Scholar]
- Motekaitis, R.J.; Martell, A.E. Halogenated Symmetrical Dipyrromethene Chelates. Inorg. Chem. 1970, 9, 1832–1839. [Google Scholar] [CrossRef]
- Clarke, E.T.; Squattrito, P.J.; Rudolf, P.R.; Motekaitis, R.Z.; Martell, A.E.; Clearfield, A. Structural Investigations of the Dipyrromethene Complexes of Calcium(II), Nickel(II) and Copper(II). Inorg. Chim. Acta 1989, 166, 221–231. [Google Scholar] [CrossRef]
- Filatov, M.A.; Lebedev, A.Y.; Mukhin, S.N.; Vinogradov, S.A.; Cheprakov, A.V. π-Extended Dipyrrins Capable of Highly Fluorogenic Complexation with Metal Ions. J. Am. Chem. Soc. 2010, 132, 9552–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corwin, A.H.; Melville, M.H. Relative Stabilities of Chelate Compounds of Pyrrole Pigments1. J. Am. Chem. Soc. 1955, 77, 2755–2759. [Google Scholar] [CrossRef]
- The First Series of Alkali Dipyrrinato Complexes. Available online: https://cdnsciencepub.com (accessed on 30 July 2022).
- Wan, W.; Silva, M.S.; McMillen, C.D.; Creager, S.E.; Smith, R.C. Highly Luminescent Heavier Main Group Analogues of Boron-Dipyrromethene. J. Am. Chem. Soc. 2019, 141, 8703–8707. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Kobayashi, K.; Nozaki, K. Tetravalent Metal Complexes as a New Family of Catalysts for Copolymerization of Epoxides with Carbon Dioxide. J. Am. Chem. Soc. 2011, 133, 10720–10723. [Google Scholar] [CrossRef]
- Thomas, K.E.; Desbois, N.; Conradie, J.; Teat, S.J.; Gros, C.P.; Ghosh, A. Gold Dipyrrin-Bisphenolates: A Combined Experimental and DFT Study of Metal–Ligand Interactions. RSC Adv. 2019, 10, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, M.; Albrecht, M.; Albrecht, M.; Nishimura, Y.; Arai, T.; Nabeshima, T. Red/Near-Infrared Luminescence Tuning of Group-14 Element Complexes of Dipyrrins Based on a Central Atom. Inorg. Chem. 2014, 53, 1355–1360. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ikeda, C.; Yamamura, M.; Nabeshima, T. Structural Interconversion and Regulation of Optical Properties of Stable Hypercoordinate Dipyrrin−Silicon Complexes. J. Am. Chem. Soc. 2011, 133, 4726–4729. [Google Scholar] [CrossRef]
- Sumiyoshi, A.; Chiba, Y.; Matsuoka, R.; Noda, T.; Nabeshima, T. Efficient Luminescent Properties and Cation Recognition Ability of Heavy Group 13 Element Complexes of N2O2- and N2O4-Type Dipyrrins. Dalton Trans. 2019, 48, 13169–13175. [Google Scholar] [CrossRef]
- Ikeda, C.; Ueda, S.; Nabeshima, T. Aluminium Complexes of N2O2-Type Dipyrrins: The First Hetero-Multinuclear Complexes of Metallo-Dipyrrins with High Fluorescencequantum Yields. Chem. Commun. 2009, 2009, 2544–2546. [Google Scholar] [CrossRef]
- Sakamoto, R.; Kusaka, S.; Kitagawa, Y.; Kishida, M.; Hayashi, M.; Takara, Y.; Tsuchiya, M.; Kakinuma, J.; Takeda, T.; Hirata, K.; et al. Fluorescent Azadipyrrinato Zinc(II) Complex: Hybridisation with a Dipyrrinato Ligand. Dalton Trans. 2012, 41, 14035–14037. [Google Scholar] [CrossRef]
- Jiang, X.-D.; Zhao, J.; Xi, D.; Yu, H.; Guan, J.; Li, S.; Sun, C.-L.; Xiao, L.-J. A New Water-Soluble Phosphorus-Dipyrromethene and Phosphorus-Azadipyrromethene Dye: PODIPY/Aza-PODIPY. Chem. Eur. J. 2015, 21, 6079–6082. [Google Scholar] [CrossRef]
- Teets, T.S.; Partyka, D.V.; Updegraff, J.B.; Gray, T.G. Homoleptic, Four-Coordinate Azadipyrromethene Complexes of D10 Zinc and Mercury. Inorg. Chem. 2008, 47, 2338–2346. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, R.M.; Robertson, K.N.; Thompson, A. Synthesis and Reactivity of Aza-Dipyrrin Alkali Metal Salts. Chem. Commun. 2018, 54, 13139–13142. [Google Scholar] [CrossRef]
- Baudron, S.A. Luminescent Metal–Organic Frameworks Based on Dipyrromethene Metal Complexes and BODIPYs. CrystEngComm 2016, 18, 4671–4680. [Google Scholar] [CrossRef]
- Stork, J.R.; Thoi, V.S.; Cohen, S.M. Rare Examples of Transition-Metal−Main-Group Metal Heterometallic Metal−Organic Frameworks from Gallium and Indium Dipyrrinato Complexes and Silver Salts: Synthesis and Framework Variability. Inorg. Chem. 2007, 46, 11213–11223. [Google Scholar] [CrossRef]
- Baudron, S.A. Dipyrrin Based Homo- and Hetero-Metallic Infinite Architectures. CrystEngComm 2010, 12, 2288–2295. [Google Scholar] [CrossRef]
- Alves, S.R.; Calori, I.R.; Tedesco, A.C. Photosensitizer-Based Metal-Organic Frameworks for Highly Effective Photodynamic Therapy. Mater. Sci. Eng. C 2021, 131, 112514. [Google Scholar] [CrossRef]
- Telfer, S.G.; Wuest, J.D. Metallotectons: Comparison of Molecular Networks Built from Racemic and Enantiomerically Pure Tris(Dipyrrinato)Cobalt(III) Complexes. Cryst. Growth Des. 2009, 9, 1923–1931. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsuda, Y.; Sakata, K.; Harada, K. Transition-Metal Complexes of Pyrrole Pigments. IX. Divalent and Trivalent Iron Chelates of Dipyrromethenes. Bull. Chem. Soc. Jpn. 1974, 47, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Manav, N.; Kesavan, P.E.; Ishida, M.; Mori, S.; Yasutake, Y.; Fukatsu, S.; Furuta, H.; Gupta, I. Phosphorescent Rhenium-Dipyrrinates: Efficient Photosensitizers for Singlet Oxygen Generation. Dalton Trans. 2019, 48, 2467–2478. [Google Scholar] [CrossRef]
- Murakami, Y.; Sakata, K.; Harada, K.; Matsuda, Y. Transition-Metal Complexes of Pyrrole Pigments. X. Divalent and Trivalent Manganese Chelates of Dipyrromethenes. Bull. Chem. Soc. Jpn. 1974, 47, 3021–3024. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Murakami, Y. Transition-Metal Complexes of Pyrrole Pigments. XIV. An Electron Spin Resonance Study of Bis(3,3′,5,5′-Tetramethyldipyrromethenato)Manganese(II). Bull. Chem. Soc. Jpn. 1977, 50, 2321–2324. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Matsuda, Y.; Iiyama, K. Transition-Metal Complexes of Pyrrole Pigments, Viii, Synthesis and Characterization of Acetatobis(3,3′,5,5′-Tetramethyldipyrromethenato)Chromium(Iii). Chem. Lett. 1972, 1, 1069–1072. [Google Scholar] [CrossRef]
- Locher, J.; Watt, F.A.; Neuba, A.G.; Schoch, R.; Munz, D.; Hohloch, S. Molybdenum(VI) Bis-Imido Complexes of Dipyrromethene Ligands. Inorg. Chem. 2020, 59, 9847–9856. [Google Scholar] [CrossRef]
- Burchinov, A.N.; Kiselev, V.M.; Penni, A.A.; Khistyaeva, V.V. Photosensitized Generation of Singlet Oxygen by Rhenium(I) Complex. Opt. Spectrosc. 2015, 119, 932–937. [Google Scholar] [CrossRef]
- Spada, R.M.; Cepeda-Plaza, M.; Gómez, M.L.; Günther, G.; Jaque, P.; Pizarro, N.; Palacios, R.E.; Vega, A. Clean Singlet Oxygen Production by a ReI Complex Embedded in a Flexible Self-Standing Polymeric Silsesquioxane Film. J. Phys. Chem. C 2015, 119, 10148–10159. [Google Scholar] [CrossRef]
- Ramos, L.D.; da Cruz, H.M.; Frin, K.P.M. Photophysical Properties of Rhenium(I) Complexes and Photosensitized Generation of Singlet Oxygen. Photochem. Photobiol. Sci. 2017, 16, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Ragone, F.; Saavedra, H.H.M.; Gara, P.M.D.; Ruiz, G.T.; Wolcan, E. Photosensitized Generation of Singlet Oxygen from Re(I) Complexes: A Photophysical Study Using LIOAS and Luminescence Techniques. J. Phys. Chem. A 2013, 117, 4428–4435. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Shafi, A.A.; Bourdelande, J.L.; Ali, S.S. Photosensitized Generation of Singlet Oxygen from Rhenium(I) and Iridium(III) Complexes. Dalton Trans. 2007, 2007, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.; Bisset, S.W.; Telfer, S.G. Postcomplexation Synthetic Routes to Dipyrrin Complexes. Dalton Trans. 2016, 45, 2440–2443. [Google Scholar] [CrossRef]
- Zhang, Y.; Crawley, M.R.; Hauke, C.E.; Friedman, A.E.; Janik, T.S.; Cook, T.R. A Bis(Dipyrrinato) Motif as a Building Block for Polynuclear Rhenium(I) Architectures. Eur. J. Inorg. Chem. 2017, 2017, 4055–4060. [Google Scholar] [CrossRef]
- Yin, H.; Stephenson, M.; Gibson, J.; Sampson, E.; Shi, G.; Sainuddin, T.; Monro, S.; McFarland, S.A. In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg. Chem. 2014, 53, 4548–4559. [Google Scholar] [CrossRef]
- Karges, J.; Blacque, O.; Goldner, P.; Chao, H.; Gasser, G. Towards Long Wavelength Absorbing Photodynamic Therapy Photosensitizers via the Extension of a [Ru(Bipy)3]2+ Core. Eur. J. Inorg. Chem. 2019, 2019, 3704–3712. [Google Scholar] [CrossRef]
- Hohlfeld, B.F.; Flanagan, K.J.; Kulak, N.; Senge, M.O.; Christmann, M.; Wiehe, A. Synthesis of Porphyrinoids, BODIPYs, and (Dipyrrinato)Ruthenium(II) Complexes from Prefunctionalized Dipyrromethanes. Eur. J. Org. Chem. 2019, 2019, 4020–4033. [Google Scholar] [CrossRef]
- Smalley, S.J.; Waterland, M.R.; Telfer, S.G. Heteroleptic Dipyrrin/Bipyridine Complexes of Ruthenium(II). Inorg. Chem. 2009, 48, 13–15. [Google Scholar] [CrossRef]
- McLean, T.M.; Cleland, D.M.; Lind, S.J.; Gordon, K.C.; Telfer, S.G.; Waterland, M.R. Strongly Absorbing π–Π* States in Heteroleptic Dipyrrin/2,2′-Bipyridine Ruthenium Complexes: Excited-State Dynamics from Resonance Raman Spectroscopy. Chem. Asian J. 2010, 5, 2036–2046. [Google Scholar] [CrossRef]
- Paitandi, R.P.; Singh, R.S.; Mukhopadhyay, S.; Sharma, G.; Koch, B.; Vishnoi, P.; Pandey, D.S. Synthesis, Characterization, DNA Binding and Cytotoxicity of Fluoro-Dipyrrin Based Arene Ruthenium(II) Complexes. Inorg. Chim. Acta 2017, 454, 117–127. [Google Scholar] [CrossRef]
- Gupta, R.K.; Kumar, A.; Paitandi, R.P.; Singh, R.S.; Mukhopadhyay, S.; Verma, S.P.; Das, P.; Pandey, D.S. Heteroleptic Arene Ru(II) Dipyrrinato Complexes: DNA, Protein Binding and Anti-Cancer Activity against the ACHN Cancer Cell Line. Dalton Trans. 2016, 45, 7163–7177. [Google Scholar] [CrossRef]
- Paitandi, R.P.; Gupta, R.K.; Singh, R.S.; Sharma, G.; Koch, B.; Pandey, D.S. Interaction of Ferrocene Appended Ru(II), Rh(III) and Ir(III) Dipyrrinato Complexes with DNA/Protein, Molecular Docking and Antitumor Activity. Eur. J. Med. Chem. 2014, 84, 17–29. [Google Scholar] [CrossRef]
- Yadav, M.; Singh, A.K.; Pandey, D.S. Heteroleptic Half-Sandwich Ru(II), Rh(III) and Ir(III) Complexes Based on 5-Ferrocenyldipyrromethene. J. Organomet. Chem. 2011, 696, 758–763. [Google Scholar] [CrossRef]
- Mabrouk, P.A.; Wrighton, M.S. Resonance Raman Spectroscopy of the Lowest Excited State of Derivatives of Tris(2,2′-Bipyridine)Ruthenium(II): Substituent Effects on Electron Localization in Mixed-Ligand Complexes. Inorg. Chem. 1986, 25, 526–531. [Google Scholar] [CrossRef]
- Telfer, S.G.; Wuest, J.D. Metallotectons: Using Enantiopure Tris(Dipyrrinato)Cobalt(III) Complexes to Build Chiral Molecular Materials. Chem. Commun. 2007, 2017, 3166–3168. [Google Scholar] [CrossRef]
- Gupta, R.K.; Pandey, R.; Sharma, G.; Prasad, R.; Koch, B.; Srikrishna, S.; Li, P.-Z.; Xu, Q.; Pandey, D.S. DNA Binding and Anti-Cancer Activity of Redox-Active Heteroleptic Piano-Stool Ru(II), Rh(III), and Ir(III) Complexes Containing 4-(2-Methoxypyridyl)Phenyldipyrromethene. Inorg. Chem. 2013, 52, 3687–3698. [Google Scholar] [CrossRef]
- Gupta, R.K.; Sharma, G.; Pandey, R.; Kumar, A.; Koch, B.; Li, P.-Z.; Xu, Q.; Pandey, D.S. DNA/Protein Binding, Molecular Docking, and in Vitro Anticancer Activity of Some Thioether-Dipyrrinato Complexes. Inorg. Chem. 2013, 52, 13984–13996. [Google Scholar] [CrossRef]
- Imberti, C.; Zhang, P.; Huang, H.; Sadler, P.J. New Designs for Phototherapeutic Transition Metal Complexes. Angew. Chem. Int. Ed. 2020, 59, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Hanson, K.; Tamayo, A.; Diev, V.V.; Whited, M.T.; Djurovich, P.I.; Thompson, M.E. Efficient Dipyrrin-Centered Phosphorescence at Room Temperature from Bis-Cyclometalated Iridium(III) Dipyrrinato Complexes. Inorg. Chem. 2010, 49, 6077–6084. [Google Scholar] [CrossRef]
- Bronner, C.; Baudron, S.A.; Hosseini, M.W. Carboxylic Acid Appended Dipyrrin for the Formation of a Hexanuclear Iridium/Copper Paddlewheel Complex. Inorg. Chem. 2010, 49, 8659–8661. [Google Scholar] [CrossRef]
- Bronner, C.; Veiga, M.; Guenet, A.; De Cola, L.; Hosseini, M.W.; Strassert, C.A.; Baudron, S.A. Excited State Properties and Energy Transfer within Dipyrrin-Based Binuclear Iridium/Platinum Dyads: The Effect of Ortho-Methylation on the Spacer. Chem. Eur. J. 2012, 18, 4041–4050. [Google Scholar] [CrossRef]
- Takaki, K.; Sakuda, E.; Ito, A.; Horiuchi, S.; Arikawa, Y.; Umakoshi, K. Controlling the Electronic Structures and Excited-State Characteristics of Dipyrrinatoiridium(III) Complexes by an Arylborane or an Arylamino Unit. Inorg. Chem. 2019, 58, 14542–14550. [Google Scholar] [CrossRef]
- Yadav, M.; Singh, A.K.; Pandey, D.S. First Examples of Heteroleptic Dipyrrin/H5-Pentamethylcyclopentadienyl Rhodium/Iridium(III) Complexes and Their Catalytic Activity. Organometallics 2009, 28, 4713–4723. [Google Scholar] [CrossRef]
- Yu, L.; Muthukumaran, K.; Sazanovich, I.V.; Kirmaier, C.; Hindin, E.; Diers, J.R.; Boyle, P.D.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Excited-State Energy-Transfer Dynamics in Self-Assembled Triads Composed of Two Porphyrins and an Intervening Bis(Dipyrrinato)Metal Complex. Inorg. Chem. 2003, 42, 6629–6647. [Google Scholar] [CrossRef]
- Sazanovich, I.V.; Kirmaier, C.; Hindin, E.; Yu, L.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Structural Control of the Excited-State Dynamics of Bis(Dipyrrinato)Zinc Complexes: Self-Assembling Chromophores for Light-Harvesting Architectures. J. Am. Chem. Soc. 2004, 126, 2664–2665. [Google Scholar] [CrossRef]
- Lee, S.; Seok, C.-H.; Park, Y.; Lee, A.; Jung, D.H.; Choi, S.-H.; Park, J. Enforced Effects of Side Group Substitution Position on Luminescence Properties; Synthesis of Bis(Dipyrrinato)Zinc Complex Derivatives. Mol. Cryst. Liquid Cryst. 2010, 531, 499327. [Google Scholar] [CrossRef]
- Trinh, C.; Kirlikovali, K.; Das, S.; Ener, M.E.; Gray, H.B.; Djurovich, P.; Bradforth, S.E.; Thompson, M.E. Symmetry-Breaking Charge Transfer of Visible Light Absorbing Systems: Zinc Dipyrrins. J. Phys. Chem. C 2014, 118, 21834–21845. [Google Scholar] [CrossRef] [Green Version]
- Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. An Extremely Bright Heteroleptic Bis(Dipyrrinato)Zinc(II) Complex. Chem. Asian J. 2012, 7, 907–910. [Google Scholar] [CrossRef]
- Alqahtani, N.Z.; Blevins, T.G.; McCusker, C.E. Quantifying Triplet State Formation in Zinc Dipyrrin Complexes. J. Phys. Chem. A 2019, 123, 10011–10018. [Google Scholar] [CrossRef]
- Mahmood, Z.; Rehmat, N.; Ji, S.; Zhao, J.; Sun, S.; Di Donato, M.; Li, M.; Teddei, M.; Huo, Y. Tuning the Triplet Excited State of Bis(Dipyrrin) Zinc(II) Complexes: Symmetry Breaking Charge Transfer Architecture with Exceptionally Long Lived Triplet State for Upconversion. Chem. Eur. J. 2020, 26, 14912–14918. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Thornbury, W.G.; Bartynski, A.N.; Thompson, M.E.; Bradforth, S.E. Manipulating Triplet Yield through Control of Symmetry-Breaking Charge Transfer. J. Phys. Chem. Lett. 2018, 9, 3264–3270. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C.; Karunaratne, V.; Rettig, S.J.; Dolphin, D. Synthesis of Meso -Phenyl-4,6-Dipyrrins, Preparation of Their Cu(II), Ni(II), and Zn(II) Chelates, and Structural Characterization of Bis[Meso-Phenyl-4,6-Dipyrrinato]Ni(II). Can. J. Chem. 1996, 74, 2182–2193. [Google Scholar] [CrossRef] [Green Version]
- Karges, J.; Blacque, O.; Chao, H.; Gasser, G. Polymeric Bis(Dipyrrinato) Zinc(II) Nanoparticles as Selective Imaging Probes for Lysosomes of Cancer Cells. Inorg. Chem. 2019, 58, 12422–12432. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Baudron, S.A.; Hosseini, M.W.; Strassert, C.A.; Guenet, A.; Cola, L.D. Dipyrrin Based Luminescent Cyclometallated Palladium and Platinum Complexes. Dalton Trans. 2009, 39, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sukhanov, A.A.; Zhao, J.; Elmali, A.; Li, X.; Dick, B.; Karatay, A.; Voronkova, V.K. Spin–Orbit Charge-Transfer Intersystem Crossing (SOCT-ISC) in Bodipy-Phenoxazine Dyads: Effect of Chromophore Orientation and Conformation Restriction on the Photophysical Properties. J. Phys. Chem. C 2019, 123, 22793–22811. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, J. Bodipy–Anthracene Dyads as Triplet Photosensitizers: Effect of Chromophore Orientation on Triplet-State Formation Efficiency and Application in Triplet–Triplet Annihilation Upconversion. Org. Lett. 2017, 19, 4492–4495. [Google Scholar] [CrossRef]
- Dance, Z.E.X.; Mickley, S.M.; Wilson, T.M.; Ricks, A.B.; Scott, A.M.; Ratner, M.A.; Wasielewski, M.R. Intersystem Crossing Mediated by Photoinduced Intramolecular Charge Transfer: Julolidine−Anthracene Molecules with Perpendicular π Systems. J. Phys. Chem. A 2008, 112, 4194–4201. [Google Scholar] [CrossRef]
- Hou, Y.; Kurganskii, I.; Elmali, A.; Zhang, H.; Gao, Y.; Lv, L.; Zhao, J.; Karatay, A.; Luo, L.; Fedin, M. Electronic Coupling and Spin–Orbit Charge Transfer Intersystem Crossing (SOCT-ISC) in Compact BDP–Carbazole Dyads with Different Mutual Orientations of the Electron Donor and Acceptor. J. Chem. Phys. 2020, 152, 114701. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Sakamoto, R.; Shimada, M.; Yamanoi, Y.; Hattori, Y.; Sugimoto, K.; Nishibori, E.; Nishihara, H. Bis(Dipyrrinato)Zinc(II) Complexes: Emission in the Solid State. Inorg. Chem. 2016, 55, 5732–5734. [Google Scholar] [CrossRef]
- Sakamoto, R.; Iwashima, T.; Kögel, J.F.; Kusaka, S.; Tsuchiya, M.; Kitagawa, Y.; Nishihara, H. Dissymmetric Bis(Dipyrrinato)Zinc(II) Complexes: Rich Variety and Bright Red to Near-Infrared Luminescence with a Large Pseudo-Stokes Shift. J. Am. Chem. Soc. 2016, 138, 5666–5677. [Google Scholar] [CrossRef]
- Mathew, D.; Hu, Y.; Zhao, J.; Arunkumar, C.; Sujatha, S. Synthesis, Structure, Photophysical Properties and Evaluation of in Vitro Cytotoxic Activity of Homoleptic Dipyrrin Based Palladium Complexes. Polyhedron 2020, 190, 114794. [Google Scholar] [CrossRef]
- Riese, S.; Holzapfel, M.; Schmiedel, A.; Gert, I.; Schmidt, D.; Würthner, F.; Lambert, C. Photoinduced Dynamics of Bis-Dipyrrinato-Palladium(II) and Porphodimethenato-Palladium(II) Complexes: Governing Near Infrared Phosphorescence by Structural Restriction. Inorg. Chem. 2018, 57, 12480–12488. [Google Scholar] [CrossRef]
- 4.1.1: Introduction to Crystal Field Theory (Octahdral Complexes). Available online: https://chem.libretexts.org/Courses/Saint_Marys_College_Notre_Dame_IN/CHEM_342%3A_Bio-inorganic_Chemistry/Readings/Week_4%3A_Ligand_Field_Theory_(Octahedral_Complexes)/4.1%3A_Ligand_Field_Theory_(LFT)_and_Crystal_Field_Theory_(CFT)_of_Octahedral_Complexes/4.1.1%3A_Introduction_to_Crystal_Field_Theory_(Octahdral_complexes) (accessed on 9 August 2022).
- Crabtree, R.H. (Ed.) The Organometallic Chemistry of the Transition Metals; Wiley GMBH: Hoboken, NJ, USA, 2019. [Google Scholar]
- March, F.C.; Couch, D.A.; Emerson, K.; Fergusson, J.E.; Robinson, W.T. Dipyrromethene Complexes of Transition Metals. Part II. Stereochemistry of Complexes of Cobalt(II), Nickel(II), Copper(II), Zinc(II), Cadmium(II), Mercury(II), and Palladium(II) and Crystal Structure Analysis of the Palladium Complex. J. Chem. Soc. A 1971, 1971, 440–448. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Miao, Q.; Shin, J.-Y.; Patrick, B.O.; Dolphin, D. Self-Assembly of Oligomeric Linear Dipyrromethene Metal Complexes. Chem. Commun. 2009, 2009, 2541–2543. [Google Scholar] [CrossRef] [Green Version]
- Guseva, G.B.; Antina, E.V.; Beresin, M.B.; V’yugin, A.I.; Nuraneeva, E.N. Preparation, Spectral and Thermal Properties of Co(II), Ni(II), Cu(II), Zn(II), and Cd(II) Complexes with Iodosubstituted 2,2′-Dipyrrolylmethene. Russ. J. Gen. Chem. 2013, 83, 1571–1579. [Google Scholar] [CrossRef]
- Brunings, K.J.; Corwin, A.H. Effect of Substituents on the Structure of the Dipyrrylmethenes. Some Relationships between the Dipyrryl- and the Triphenylmethane Dyes1,2. J. Am. Chem. Soc. 1944, 66, 337–342. [Google Scholar] [CrossRef]
- Gill, H.S.; Finger, I.; Božidarević, I.; Szydlo, F.; Scott, M.J. Preparation of α,β-Unsubstituted Meso-Arylbidipyrrins via Metal-Templated, Oxidative Coupling of Dipyrrins. New J. Chem. 2005, 29, 68–71. [Google Scholar] [CrossRef]
- Cotton, F.A.; DeBoer, B.G.; Pipal, J.R. Crystal and Molecular Structure of Bis(3,3′,5,5′-Tetramethyldipyrromethenato)Nickel(II). Inorg. Chem. 1970, 9, 783–788. [Google Scholar] [CrossRef]
- Murakami, Y.; Sakata, K. Transition-Metal Complexes of Pyrrole Pigments. I. Electronic and Vibrational Spectra of Cobalt(II), Nickel(II) and Copper(II) Complexes of Some Dipyrromethenes. Inorg. Chim. Acta 1968, 2, 273–279. [Google Scholar] [CrossRef]
- Fergusson, J.E.; Ramsay, C.A. 971. Dipyrromethene Complexes of Transition Metals. Part I. Tetrahedral Complexes of Cobalt(II), Nickel(II), Copper(II), and Zinc(II). J. Chem. Soc. 1965, 1965, 5222–5225. [Google Scholar] [CrossRef]
- Jutzi, P. Advanced Inorganic Chemistry. Von F. A. Cotton und G. Wilkinson. 5. Aufl. John Wiley & Sons, New York 1988. 1455 S., Tab., Abb., geb. £ 21.95. ISBN 0-471849979. Chem. Unserer Zeit 1989, 23, 741–754. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, Z.; Dong, Y.; Zhu, W. Synthesis and Electrochemistry of Meso-Substitutes Dipyrromethene Nickel (II) Complexes. Polyhedron 2015, 102, 578–582. [Google Scholar] [CrossRef]
- Xue, Z.; Dong, Y.; Ma, J.; Wang, Y.; Zhu, W. Synthesis, Characterization and Electrochemistry of Dipyrrinato Nickel(II) Complexes with Different Aromatic Rings to Meso-Position. Polyhedron 2017, 127, 287–292. [Google Scholar] [CrossRef]
- Elder, M.; Penfold, B.R. Crystal Structure of Bis(Dipyrromethene)Copper(II). J. Chem. Soc. A 1969, 1969, 2556–2559. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsuda, Y.; Sakata, K. Transition Metal Complexes of Pyrrole Pigments. IV. Electronic and Vibrational Spectra of Cobalt(II), Nickel(II), and Copper(II) Complexes of Some Substituted Dipyrromethenes. Inorg. Chem. 1971, 10, 1728–1734. [Google Scholar] [CrossRef]
- Servaty, K.; Cauët, E.; Thomas, F.; Lambermont, J.; Gerbaux, P.; Winter, J.D.; Ovaere, M.; Volker, L.; Vaeck, N.; Meervelt, L.V.; et al. Peculiar Properties of Homoleptic Cu Complexes with Dipyrromethene Derivatives. Dalton Trans. 2013, 42, 14188–14199. [Google Scholar] [CrossRef]
- Toyoda, R.; Tsuchiya, M.; Sakamoto, R.; Matsuoka, R.; Wu, K.-H.; Hattori, Y.; Nishihara, H. Heteroleptic Bis(Dipyrrinato)Copper(II) and Nickel(II) Complexes. Dalton Trans. 2015, 44, 15103–15106. [Google Scholar] [CrossRef]
- Guseva, G.B.; Antina, E.V.; Berezin, M.B.; V’yugin, A.I. Influence of Metal Cation on Chromophore Properties of Complexes of Some d Metals with α,α-Dipyrrolylmethene. Russ. J. Gen. Chem. 2004, 74, 1282–1285. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. 637. The Stability of Transition-Metal Complexes. J. Chem. Soc. 1953, 1953, 3192–3210. [Google Scholar] [CrossRef]
- Brückner, C.; Zhang, Y.; Rettig, S.J.; Dolphin, D. Synthesis, Derivatization and Structural Characterization of Octahedral Tris(5-Phenyl-4,6-Dipyrrinato) Complexes of Cobalt(III) and Iron(III). Inorg. Chim. Acta 1997, 263, 279–286. [Google Scholar] [CrossRef]
- Das, S.; Gupta, I. Triphenylamine Substituted Dipyrrinato Metal Complexes: Synthesis, Optical and Electrochemical Studies. Inorg. Chem. Commun. 2015, 60, 54–60. [Google Scholar] [CrossRef]
- Ostadhosseini, N.; Shamlouei, H.R.; Bahrami, H. TDDFT Study of the Influence of C20 Fullerene on Optical Properties of BODIPY and Two Its Analogs: AlDIPY, GaDIPY. J. Inorg. Organomet. Polym. 2020, 30, 4160–4169. [Google Scholar] [CrossRef]
- Kusaka, S.; Sakamoto, R.; Nishihara, H. Luminescent Heteroleptic Tris(Dipyrrinato)Indium(III) Complexes. Inorg. Chem. 2014, 53, 3275–3277. [Google Scholar] [CrossRef]
- Gianopoulos, C.G.; Kirschbaum, K.; Mason, M.R. Mono- and Bimetallic Aluminum Alkyl, Aryl, and Hydride Complexes of a Bulky Dipyrromethene Ligand. Organometallics 2014, 33, 4503–4511. [Google Scholar] [CrossRef]
- Gianopoulos, C.G.; Kumar, N.; Zhao, Y.; Jia, L.; Kirschbaum, K.; Mason, M.R. Aluminum Alkoxide, Amide and Halide Complexes Supported by a Bulky Dipyrromethene Ligand: Synthesis, Characterization, and Preliminary ε-Caprolactone Polymerization Activity. Dalton Trans. 2016, 45, 13787–13797. [Google Scholar] [CrossRef]
- Hsieh, A.T.T.; Rogers, C.A.; West, B.O. Synthesis, Properties and Long Range 203,205Tl-13C Couplings in the 13C N.M.R. Spectrum of an Unusual Dimethylthallium(III) Complex of 4,4’-Diethoxycarbonyl-3,3′,5,5′-Tetramethyldipyrromethene. Aust. J. Chem. 1976, 29, 49–54. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kushida, T.; Kawashima, T. Synthesis and Reversible Control of the Fluorescent Properties of a Divalent Tin Dipyrromethene. J. Am. Chem. Soc. 2009, 131, 10836–10837. [Google Scholar] [CrossRef]
- Sharma, R.; Ghosh, A.; Wolfram, B.; Bröring, M.; Ravikanth, M. Synthesis and Characterization of Hexa-Coordinated Sn(IV) Complexes of Meso-Aryl Dipyrrins. Dalton Trans. 2013, 42, 5627–5630. [Google Scholar] [CrossRef]
- Kämpfe, A.; Kroke, E.; Wagler, J. Silicon Compounds of 1,1-Bis(Pyrrol-2-Yl)Ethenes: Molecular Structures and Chemical and Spectroscopic Properties. Organometallics 2014, 33, 112–120. [Google Scholar] [CrossRef]
- Fihey, A.; Favennec, A.; Guennic, B.L.; Jacquemin, D. Investigating the Properties of PODIPYs (Phosphorus-Dipyrromethene) with Ab Initio Tools. Phys. Chem. Chem. Phys. 2016, 18, 9358–9366. [Google Scholar] [CrossRef]
- Jiang, X.-D.; Yu, H.-F.; Zhao, J.-L.; Sun, C.-L.; Xie, Y.; Xiao, L.-J. A Colorimetric Chemosensor Based on New Water-Soluble PODIPY Dye for Hg2+ Detection. Chin. Chem. Lett. 2015, 26, 1241–1245. [Google Scholar] [CrossRef]
- Madhu, S.; Kalaiyarasi, R.; Basu, S.K.; Jadhav, S.; Ravikanth, M. A Boron-Dipyrrin–Mercury(II) Complex as a Fluorescence Turn-on Sensor for Chloride and Applications towards Logic Gates. J. Mater. Chem. C 2014, 2, 2534–2544. [Google Scholar] [CrossRef]
- Gresser, R.; Hoyer, A.; Hummert, M.; Hartmann, H.; Leo, K.; Riede, M. Homoleptic Co(II), Ni(II), Cu(II), Zn(II) and Hg(II) Complexes of Bis-(Phenyl)-Diisoindol-Aza-Methene. Dalton Trans. 2011, 40, 3476–3483. [Google Scholar] [CrossRef] [Green Version]
- Béziau, A.; Baudron, S.A.; Guenet, A.; Hosseini, M.W. Luminescent Coordination Polymers Based on Self-Assembled Cadmium Dipyrrin Complexes. Chem. Eur. J. 2013, 19, 3215–3223. [Google Scholar] [CrossRef]
- Antina, L.A.; Dudina, N.A.; Guseva, G.B.; Berezin, M.B.; V’yugin, A.I. Synthesis and Photophysical Properties of Cd(II) and Cu(II) Complexes with Decamethylated Bis(Dipyrrolylmethene). Russ. J. Gen. Chem. 2011, 81, 2349–2351. [Google Scholar] [CrossRef]
- Porter, C.R. 74. The Stereochemistry of Metallic Derivatives of Pyrromethenes. J. Chem. Soc. 1938, 1938, 368–372. [Google Scholar] [CrossRef]
- Vidal, F.; Jäkle, F. Functional Polymeric Materials Based on Main-Group Elements. Angew. Chem. Int. Ed. 2019, 58, 5846–5870. [Google Scholar] [CrossRef]
- Alahmadi, A.F.; Lalancette, R.A.; Jäkle, F. Highly Luminescent Ladderized Fluorene Copolymers Based on B–N Lewis Pair Functionalization. Macromol. Rap. Commun. 2018, 39, 1800456. [Google Scholar] [CrossRef]
- Wright, V.A.; Gates, D.P. Poly(p-Phenylenephosphaalkene): A π-Conjugated Macromolecule Containing P=C Bonds in the Main Chain. Angew. Chem. Int. Ed. 2002, 41, 2389–2392. [Google Scholar] [CrossRef]
- Smith, R.C.; Protasiewicz, J.D. Conjugated Polymers Featuring Heavier Main Group Element Multiple Bonds: A Diphosphene-PPV. J. Am. Chem. Soc. 2004, 126, 2268–2269. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. (Eds.) 7—Aluminium, Gallium, Indium and Thallium. In Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1997; pp. 216–267. ISBN 978-0-7506-3365-9. [Google Scholar]
- Igbokwe, I.O.; Igwenagu, E.; Igbokwe, N.A. Aluminium Toxicosis: A Review of Toxic Actions and Effects. Interdiscip. Toxicol. 2019, 12, 45–70. [Google Scholar] [CrossRef] [Green Version]
- Willhite, C.C.; Karyakina, N.A.; Yokel, R.A.; Yenugadhati, N.; Wisniewski, T.M.; Arnold, I.M.F.; Momoli, F.; Krewski, D. Systematic Review of Potential Health Risks Posed by Pharmaceutical, Occupational and Consumer Exposures to Metallic and Nanoscale Aluminum, Aluminum Oxides, Aluminum Hydroxide and Its Soluble Salts. Crit. Rev. Toxicol. 2014, 44, 934439. [Google Scholar] [CrossRef]
- Kudinova, N.V.; Berezov, T.T. Photodynamic Therapy of Cancer: Search for Ideal Photosensitizer. Biochem. Mosc. Suppl. Ser. B 2010, 4, 95–103. [Google Scholar] [CrossRef]
- Kim, H.; Burghart, A.; Welch, M.B.; Reibenspies, J.; Burgess, K. Synthesis and Spectroscopic Properties of a New 4-Bora-3a,4a-Diaza-s-Indacene (BODIPY®) Dye. Chem. Commun. 1999, 1999, 1889–1890. [Google Scholar] [CrossRef]
- Wang, S. Luminescence and Electroluminescence of Al(III), B(III), Be(II) and Zn(II) Complexes with Nitrogen Donors. Coord. Chem. Rev. 2001, 215, 79–98. [Google Scholar] [CrossRef]
- Saikawa, M.; Daicho, M.; Nakamura, T.; Uchida, J.; Yamamura, M.; Nabeshima, T. Synthesis of a New Family of Ionophores Based on Aluminum–Dipyrrin Complexes (ALDIPYs) and Their Strong Recognition of Alkaline Earth Ions. Chem. Commun. 2016, 52, 4014–4017. [Google Scholar] [CrossRef]
- Toguchi, S.; Ishikawa, H.; Morioka, Y.; Oda, A. Organic Electroluminescence Device. U.S. Patent 6,759,144, 6 July 2004. [Google Scholar]
- Karges, J.; Gasser, G. Synthesis, Characterisation and Biological Evaluation of π-Extended Fe(II) Bipyridine Complexes as Potential Photosensitizers for Photodynamic Therapy. Inorg. Chim. Acta 2020, 499, 119196. [Google Scholar] [CrossRef]
- Karges, J.; Goldner, P.; Gasser, G. Synthesis, Characterization, and Biological Evaluation of Red-Absorbing Fe(II) Polypyridine Complexes. Inorganics 2019, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Karges, J.; Xiong, K.; Blacque, O.; Chao, H.; Gasser, G. Highly Cytotoxic Copper(II) Terpyridine Complexes as Anticancer Drug Candidates. Inorg. Chim. Acta 2021, 516, 120137. [Google Scholar] [CrossRef]
- Buglak, A.A.; Charisiadis, A.; Sheehan, A.; Kingsbury, C.J.; Senge, M.O.; Filatov, M.A. Quantitative Structure-Property Relationship Modelling for the Prediction of Singlet Oxygen Generation by Heavy-Atom-Free BODIPY Photosensitizers. Chem. Eur. J. 2021, 27, 9934–9947. [Google Scholar] [CrossRef] [PubMed]




























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teeuwen, P.C.P.; Melissari, Z.; Senge, M.O.; Williams, R.M. Metal Coordination Effects on the Photophysics of Dipyrrinato Photosensitizers. Molecules 2022, 27, 6967. https://doi.org/10.3390/molecules27206967
Teeuwen PCP, Melissari Z, Senge MO, Williams RM. Metal Coordination Effects on the Photophysics of Dipyrrinato Photosensitizers. Molecules. 2022; 27(20):6967. https://doi.org/10.3390/molecules27206967
Chicago/Turabian StyleTeeuwen, Paula C. P., Zoi Melissari, Mathias O. Senge, and René M. Williams. 2022. "Metal Coordination Effects on the Photophysics of Dipyrrinato Photosensitizers" Molecules 27, no. 20: 6967. https://doi.org/10.3390/molecules27206967