
Synthesis of Trifluoromethylated Monoterpene Amino Alcohols
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
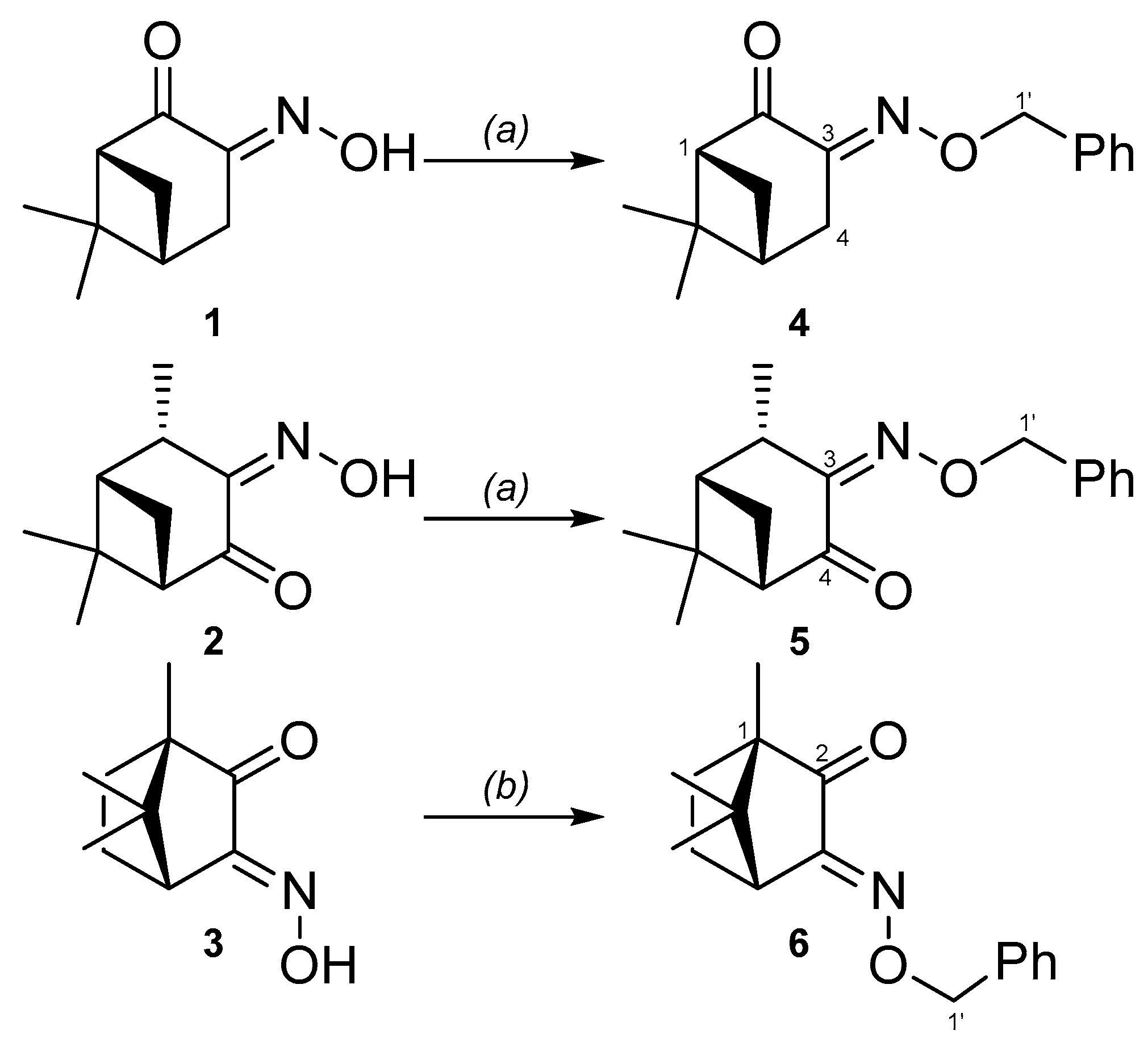
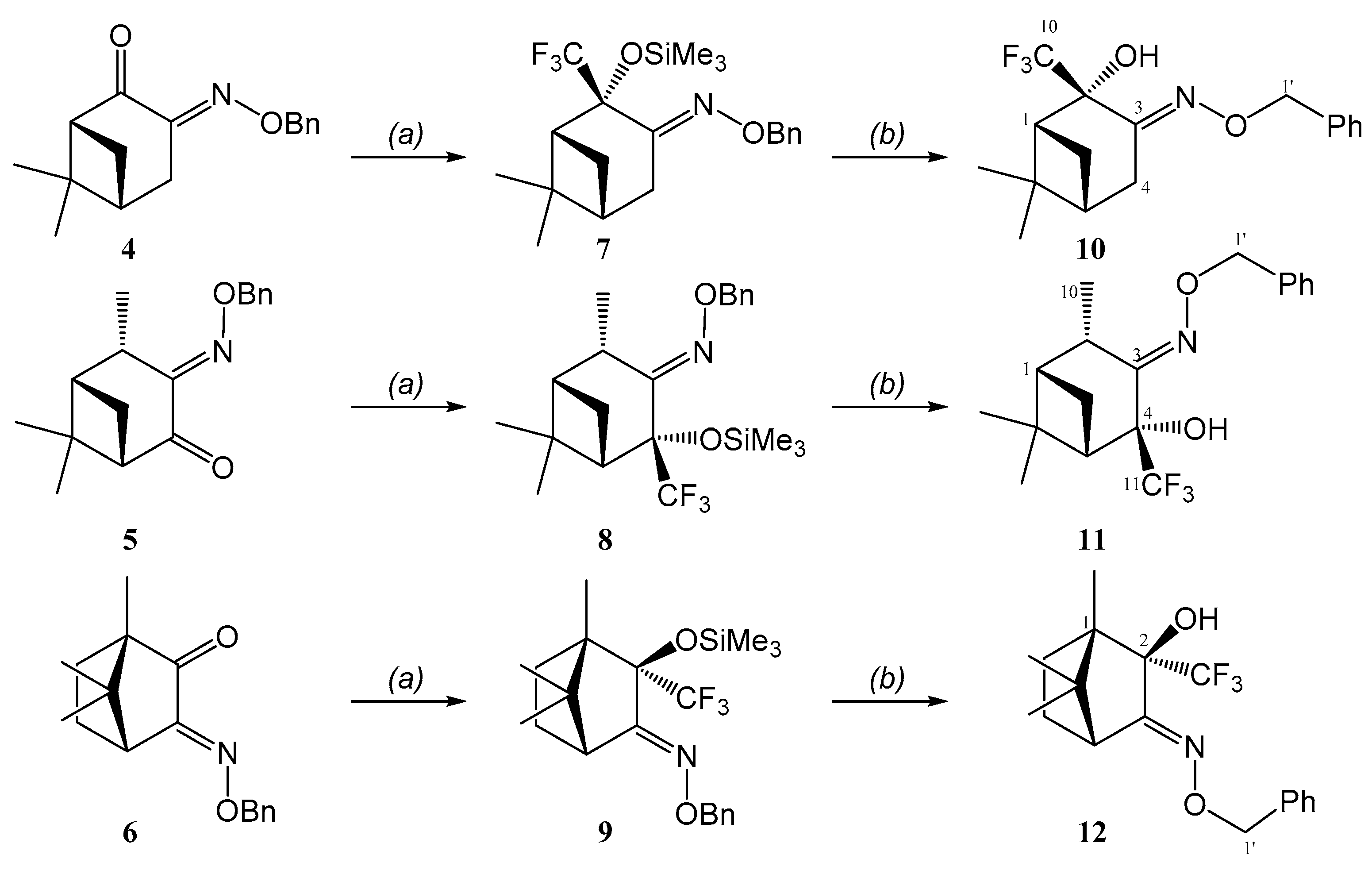
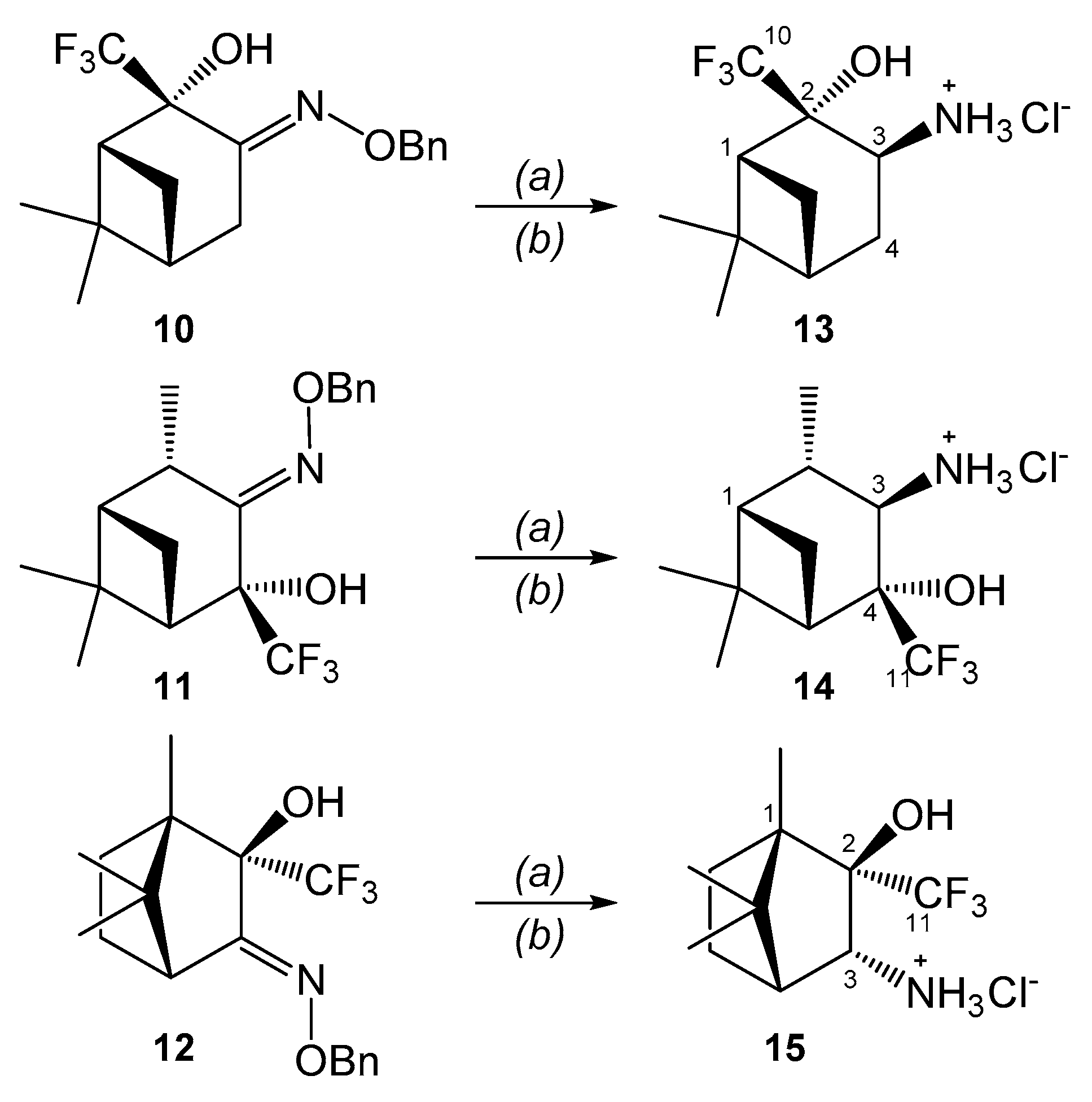
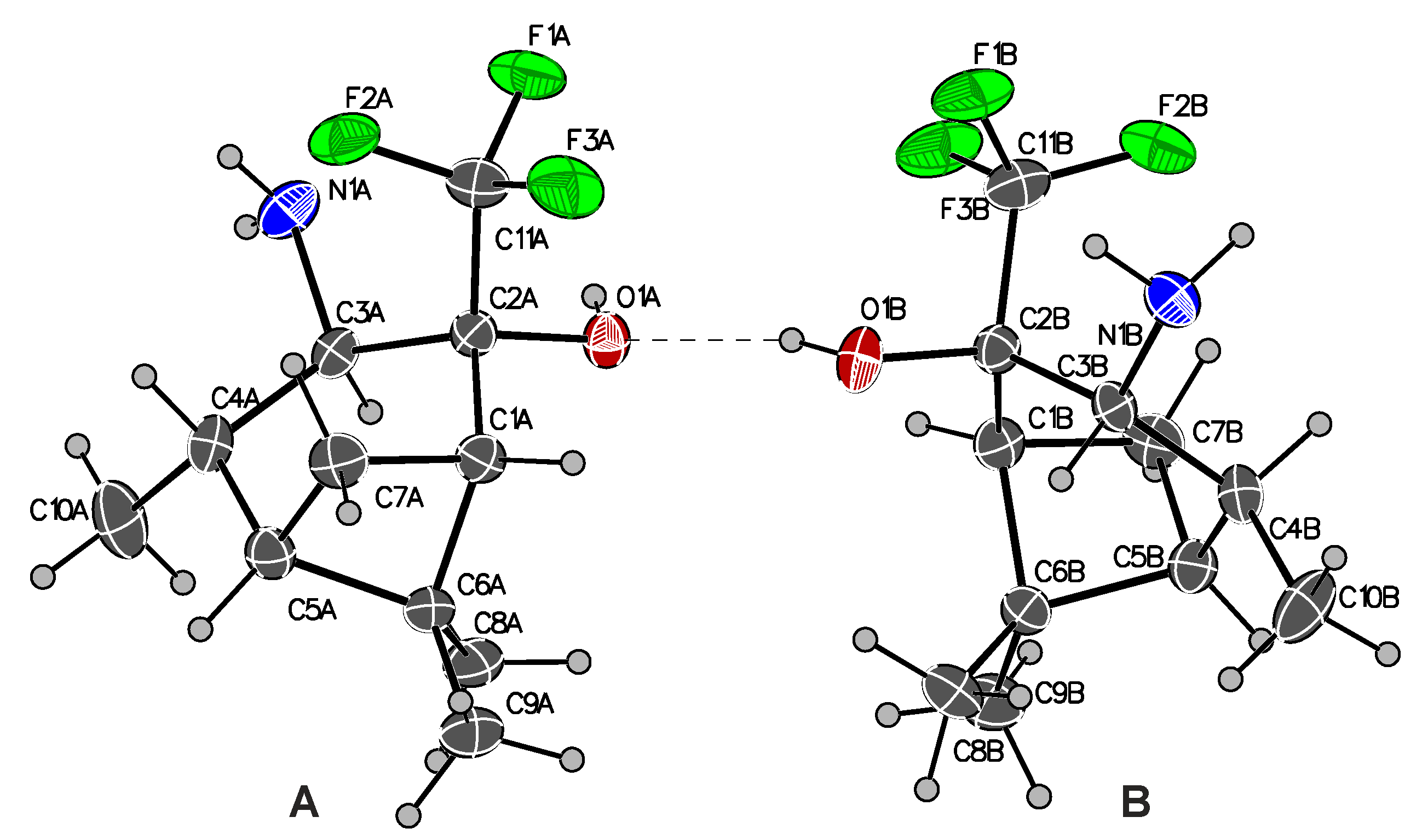
2. Results
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Benzyl-O-Oximes 4 and 5
3.3. General Procedure for Trifluoromethylation of β-Keto-Benzyl-O-Oximes 4–6
3.4. General Procedure for the Preparation of Trifluoromethylated Amino Alcohols 13–15
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nikitina, L.E.; Artemova, N.P.; Startseva, V.A. Natural and Thiomodified Monoterpenoids (Russian Edition); LAP Lambert: Saarbrücken, Germany, 2011; ISBN 978-3-8484-3023-9. [Google Scholar]
- Sikkema, J.; de Bont, J.A.; Poolman, B. Mechanisms of membrane toxicity of hydrocarbons. Microbiol. Rev. 1995, 59, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Uribe, S.; Pena, A. Toxicity of allelopathic monoterpene suspensions on yeast dependence on droplet size. J. Chem. Ecol. 1990, 16, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.P. Roles of Fluorine in Drug Design and Drug Action. Lett. Drug Des. Discov. 2019, 16, 1089–1109. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Wang, M.; Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev. 2015, 115, 683–730. [Google Scholar] [CrossRef]
- Johnson, B.M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Yoder, N.C.; Kumar, K. Fluorinated amino acids in protein design and engineering. Chem. Soc. Rev. 2002, 31, 335–341. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Qiu, X.; Qing, F. Recent Advances in the Synthesis of Fluorinated Amino Acids. Eur. J. Org. Chem. 2011, 2011, 3261–3278. [Google Scholar] [CrossRef]
- Ibn El Alami, M.S.; El Amrani, M.A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chiral Ligands Derived from Monoterpenes: Application in the Synthesis of Optically Pure Secondary Alcohols via Asymmetric Catalysis. Chem. -A Eur. J. 2015, 21, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Reddy, U.V.S.; Chennapuram, M.; Seki, C.; Kwon, E.; Okuyama, Y.; Nakano, H. Catalytic Efficiency of Primary β-Amino Alcohols and Their Derivatives in Organocatalysis. Eur. J. Org. Chem. 2016, 2016, 4124–4143. [Google Scholar] [CrossRef]
- Nakano, H.; Owolabi, I.A.; Chennapuram, M.; Okuyama, Y.; Kwon, E.; Seki, C.; Tokiwa, M.; Takeshita, M. β-Amino Alcohol Organocatalysts for Asymmetric Additions. Heterocycles 2018, 97, 647–667. [Google Scholar] [CrossRef]
- Siyutkin, D.E.; Kucherenko, A.S.; Frolova, L.L.; Kuchin, A.V.; Zlotin, S.G. N-Pyrrolidine-2-ylmethyl)-2-hydroxy-3-aminopinanes as novel organocatalysts for asymmetric conjugate additions of ketones to α-nitroalkenes. Tetrahedron Asymmetry 2013, 24, 776–779. [Google Scholar] [CrossRef]
- Banina, O.A.; Sudarikov, D.V.; Nigmatov, A.G.; Frolova, L.L.; Slepukhin, P.A.; Zlotin, S.G.; Kutchin, A.V. Carane amino alcohols as organocatalysts in asymmetric aldol reaction of isatin with acetone. Russ. Chem. Bull. 2017, 66, 293–296. [Google Scholar] [CrossRef]
- Frolova, L.L.; Sudarikov, D.V.; Alekseev, I.N.; Banina, O.A.; Slepukhin, P.A.; Kutchin, A.V. Synthesis of new enantiomerically pure β-amino alcohols of the pinane series. Russ. J. Org. Chem. 2017, 53, 335–343. [Google Scholar] [CrossRef]
- Zlotin, S.G.; Banina, O.A.; Sudarikov, D.V.; Nigmatov, A.G.; Frolova, L.L.; Kutchin, A.V. Asymmetric aldol reaction of isatins with acetone in the presence of terpene amino alcohols. Mendeleev Commun. 2020, 30, 147–149. [Google Scholar] [CrossRef]
- Fache, F. Asymmetric fluorous catalysis: The particular case of nitrogen-containing chiral auxiliaries. New J. Chem. 2004, 28, 1277–1283. [Google Scholar] [CrossRef]
- Uneyama, K. Organofluorine Chemistry; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2008; ISBN 1-4051-7293-2. [Google Scholar]
- Harada, A.; Fujiwara, Y.; Katagiri, T. Improvement of the asymmetry-inducing ability of a trifluoromethylated amino alcohol by electron donation to a CF3 group. Tetrahedron Asymmetry 2008, 19, 1210–1214. [Google Scholar] [CrossRef]
- Mlostoń, G.; Obijalska, E.; Heimgartner, H. Synthesis of β-amino-α-trifluoromethyl alcohols and their applications in organic synthesis. J. Fluor. Chem. 2010, 131, 829–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-H.; Qiu, X.-L.; Qing, F.-L. Synthesis and utilization of trifluoromethylated amino alcohol ligands for the enantioselective Reformatsky reaction and addition of diethylzinc to N-(diphenylphosphinoyl)imine. Tetrahedron 2008, 64, 7353–7361. [Google Scholar] [CrossRef]
- Dahanayake, J.N.; Kasireddy, C.; Karnes, J.P.; Verma, R.; Steinert, R.M.; Hildebrandt, D.; Hull, O.A.; Ellis, J.M.; Mitchell-Koch, K.R. Progress in Our Understanding of 19F Chemical Shifts. In Annual Reports on NMR Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2018; Volume 93, pp. 281–365. ISBN 978-0-12-814913-3. [Google Scholar]
- Howe, P.W. Recent developments in the use of fluorine NMR in synthesis and characterisation. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Vulpetti, A. Fluorine-Protein Interactions and 19F NMR Isotropic Chemical Shifts: An Empirical Correlation with Implications for Drug Design. ChemMedChem 2011, 6, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Gee, C.T.; Arntson, K.E.; Urick, A.K.; Mishra, N.K.; Hawk, L.M.L.; Wisniewski, A.J.; Pomerantz, W.C.K. Protein-observed 19F-NMR for fragment screening, affinity quantification and druggability assessment. Nat. Protoc. 2016, 11, 1414–1427. [Google Scholar] [CrossRef]
- Reich, R.M.; Kaposi, M.; Pöthig, A.; Kühn, F.E. Kinetic studies of fluorinated aryl molybdenum(ii) tricarbonyl precursors in epoxidation catalysis. Catal. Sci. Technol. 2016, 6, 4970–4977. [Google Scholar] [CrossRef] [Green Version]
- Zientek, N.; Laurain, C.; Meyer, K.; Paul, A.; Engel, D.; Guthausen, G.; Kraume, M.; Maiwald, M. Automated data evaluation and modelling of simultaneous 19 F-1 H medium-resolution NMR spectra for online reaction monitoring. Org. Magn. Reson. 2016, 54, 513–520. [Google Scholar] [CrossRef]
- Weidener, D.; Singh, K.; Blümich, B. Synthesis of α-fluoro-α,β-unsaturated esters monitored by 1D and 2D benchtop NMR spectroscopy. Org. Magn. Reson. 2019, 57, 852–860. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Hu, Y.; Li, Y.; Yang, Y.; Zha, Z.; Wang, Z. Highly Enantioselective Construction of Fluoroalkylated Quaternary Stereocenters via Organocatalytic Dehydrated Mannich Reaction of Unprotected Hemiaminals with Ketones. Org. Lett. 2015, 17, 5036–5039. [Google Scholar] [CrossRef]
- Ibba, F.; Pupo, G.; Thompson, A.L.; Brown, J.M.; Claridge, T.D.W.; Gouverneur, V. Impact of Multiple Hydrogen Bonds with Fluoride on Catalysis: Insight from NMR Spectroscopy. J. Am. Chem. Soc. 2020, 142, 19731–19744. [Google Scholar] [CrossRef]
- Zhao, Y.; Swager, T.M. Simultaneous Chirality Sensing of Multiple Amines by 19F NMR. J. Am. Chem. Soc. 2015, 137, 3221–3224. [Google Scholar] [CrossRef]
- Jang, S.; Park, H.; Duong, Q.H.; Kwahk, E.-J.; Kim, H. Determining the Enantiomeric Excess and Absolute Configuration of In Situ Fluorine-Labeled Amines and Alcohols by 19F NMR Spectroscopy. Anal. Chem. 2021, 94, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, X.; Bian, G.; Song, L. A chiral sensor for recognition of varied amines based on 19F NMR signals of newly designed rhodium complexes. Chem. Commun. 2019, 55, 6098–6101. [Google Scholar] [CrossRef] [PubMed]
- Apparu, M.; Ben Tiba, Y.; Léo, P.-M.; Hamman, S.; Coulombeau, C. Determination of the enantiomeric purity and the configuration of β-aminoalcohols using (R)-2-fluorophenylacetic acid (AFPA) and fluorine-19 NMR: Application to β-blockers. Tetrahedron Asymmetry 2000, 11, 2885–2898. [Google Scholar] [CrossRef]
- Sabot, C.; Mosser, M.; Antheaume, C.; Mioskowski, C.; Baati, R.; Wagner, A. Novel chiral derivatizing isothiocyanate-based agent for the enantiomeric excess determination of amines. Chem. Commun. 2009, 23, 3410–3412. [Google Scholar] [CrossRef]
- Binder, C.M.; Bautista, A.; Zaidlewicz, M.; Krzemiński, M.P.; Oliver, A.; Singaram, B. Dual Stereoselectivity in the Dialkylzinc Reaction Using (−)-β-Pinene Derived Amino Alcohol Chiral Auxiliaries. J. Org. Chem. 2009, 74, 2337–2343. [Google Scholar] [CrossRef]
- Chuo, T.H.; Boobalan, R.; Chen, C. Camphor-Based Schiff Base Of 3-Endo-Aminoborneol (SBAB): Novel Ligand for Vanadium-Catalyzed Asymmetric Sulfoxidation and Subsequent Kinetic Resolution. ChemistrySelect 2016, 1, 2174–2180. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Mandal, M.; Olah, G.A. Asymmetric Synthesis of Trifluoromethylated Allylic Amines Using α,β-Unsaturated N-tert-Butanesulfinimines. Org. Lett. 2001, 3, 2847–2850. [Google Scholar] [CrossRef]
- Petrov, V.A.; Davidson, F.; Marshall, W. Reactions of quadricyclane with fluorinated nitrogen-containing compounds. Synthesis of 3-aza-4-perfluoroalkyl-tricyclo [4.2.1.02,5]non-3,7-dienes. J. Fluor. Chem. 2004, 125, 1621–1628. [Google Scholar] [CrossRef]
- Radchenko, D.S.; Michurin, O.M.; Chernykh, A.V.; Lukin, O.; Mykhailiuk, P.K. An easy synthesis of α-trifluoromethyl-amines from aldehydes or ketones using the Ruppert-Prakash reagent. Tetrahedron Lett. 2013, 54, 1897–1898. [Google Scholar] [CrossRef]
- Sudarikov, D.V.; Krymskaya, Y.V.; Il’Chenko, N.O.; Slepukhin, P.A.; Rubtsova, S.A.; Kutchin, A.V. Synthesis and biological activity of fluorine-containing amino derivatives based on 4-caranethiol. Russ. Chem. Bull. 2018, 67, 731–742. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Yudin, A.K. Perfluoroalkylation with Organosilicon Reagents. Chem. Rev. 1997, 97, 757–786. [Google Scholar] [CrossRef]
- Jaworska, M.; Pawlak, T.; Kruszyński, R.; Ćwiklińska, M.; Krzemiński, M. NMR Crystallography Comparative Studies of Chiral (1R,2S,3R,5R)-3-Amino-6,6-dimethylbicyclo [3.1.1]heptan-2-ol and Its p-Toluenesulfonamide Derivative. Cryst. Growth Des. 2012, 12, 5956–5965. [Google Scholar] [CrossRef]
- SAINT. Data Reduction and Correction Program; Bruker AXS: Fitchburg, WI, USA, 2014. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, P.A.; Sudarikov, D.V.; Frolova, L.L.; Rumyantcev, R.V.; Rubtsova, S.A.; Kutchin, A.V. Synthesis of Trifluoromethylated Monoterpene Amino Alcohols. Molecules 2022, 27, 7068. https://doi.org/10.3390/molecules27207068
Petrova PA, Sudarikov DV, Frolova LL, Rumyantcev RV, Rubtsova SA, Kutchin AV. Synthesis of Trifluoromethylated Monoterpene Amino Alcohols. Molecules. 2022; 27(20):7068. https://doi.org/10.3390/molecules27207068
Chicago/Turabian StylePetrova, Polina A., Denis V. Sudarikov, Larisa L. Frolova, Roman V. Rumyantcev, Svetlana A. Rubtsova, and Aleksandr V. Kutchin. 2022. "Synthesis of Trifluoromethylated Monoterpene Amino Alcohols" Molecules 27, no. 20: 7068. https://doi.org/10.3390/molecules27207068