
Classical vs. Non-Classical Cyclometalated Pt(II) Complexes
 , , ,
, , ,
Abstract
:
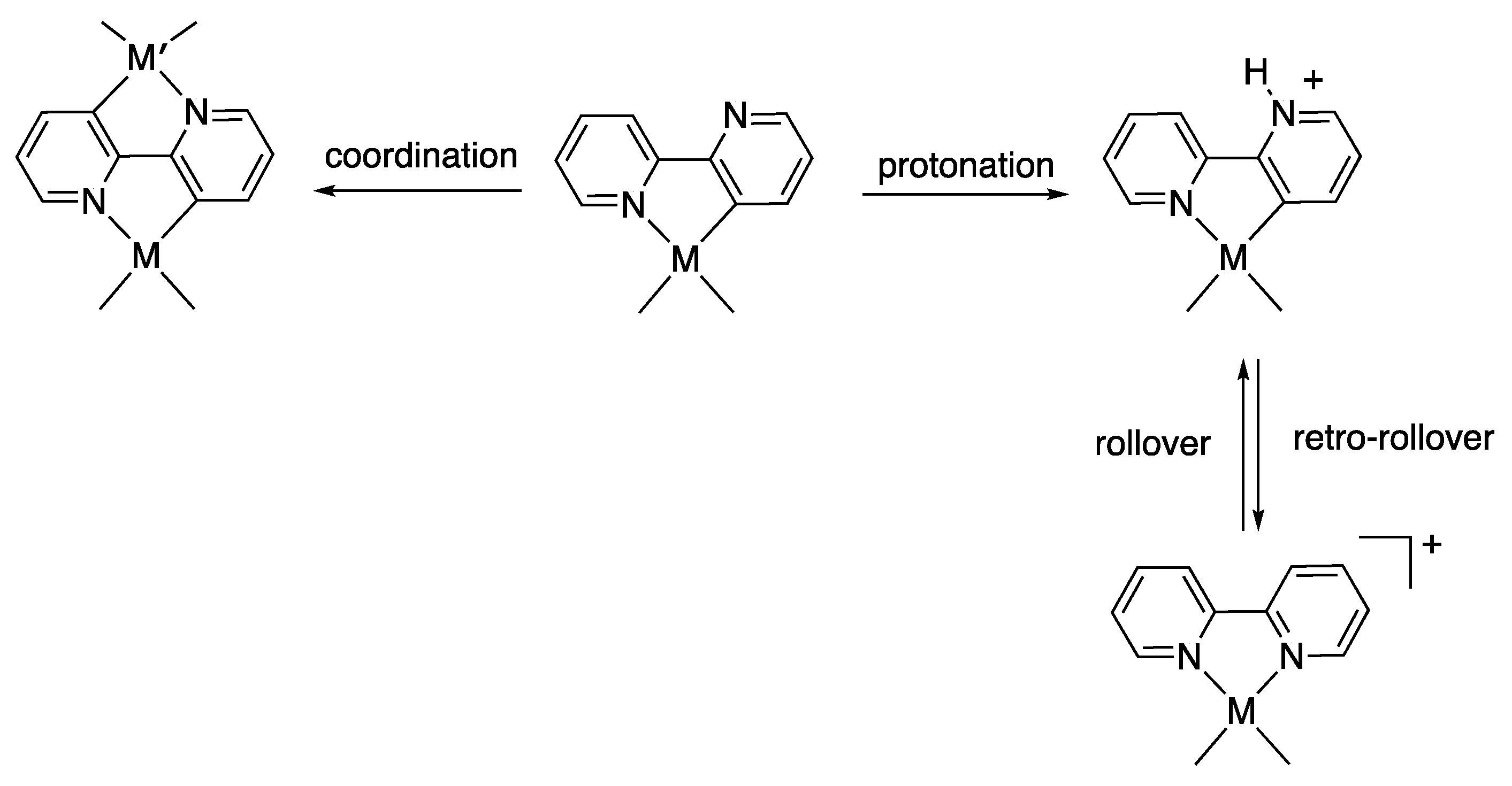
1. Introduction
2. Results and Discussion
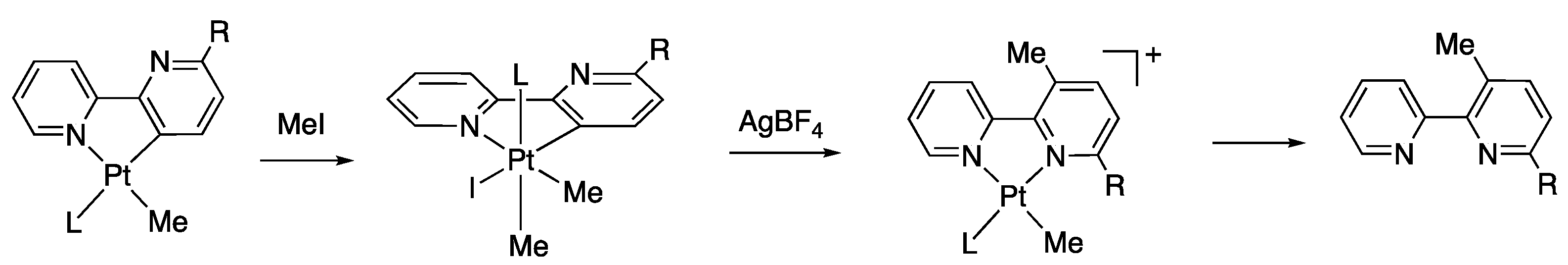
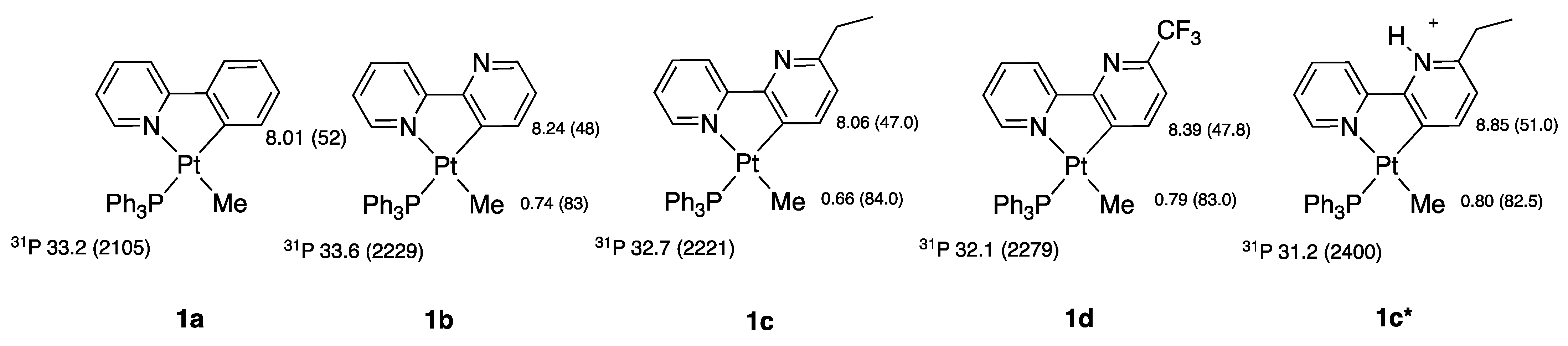
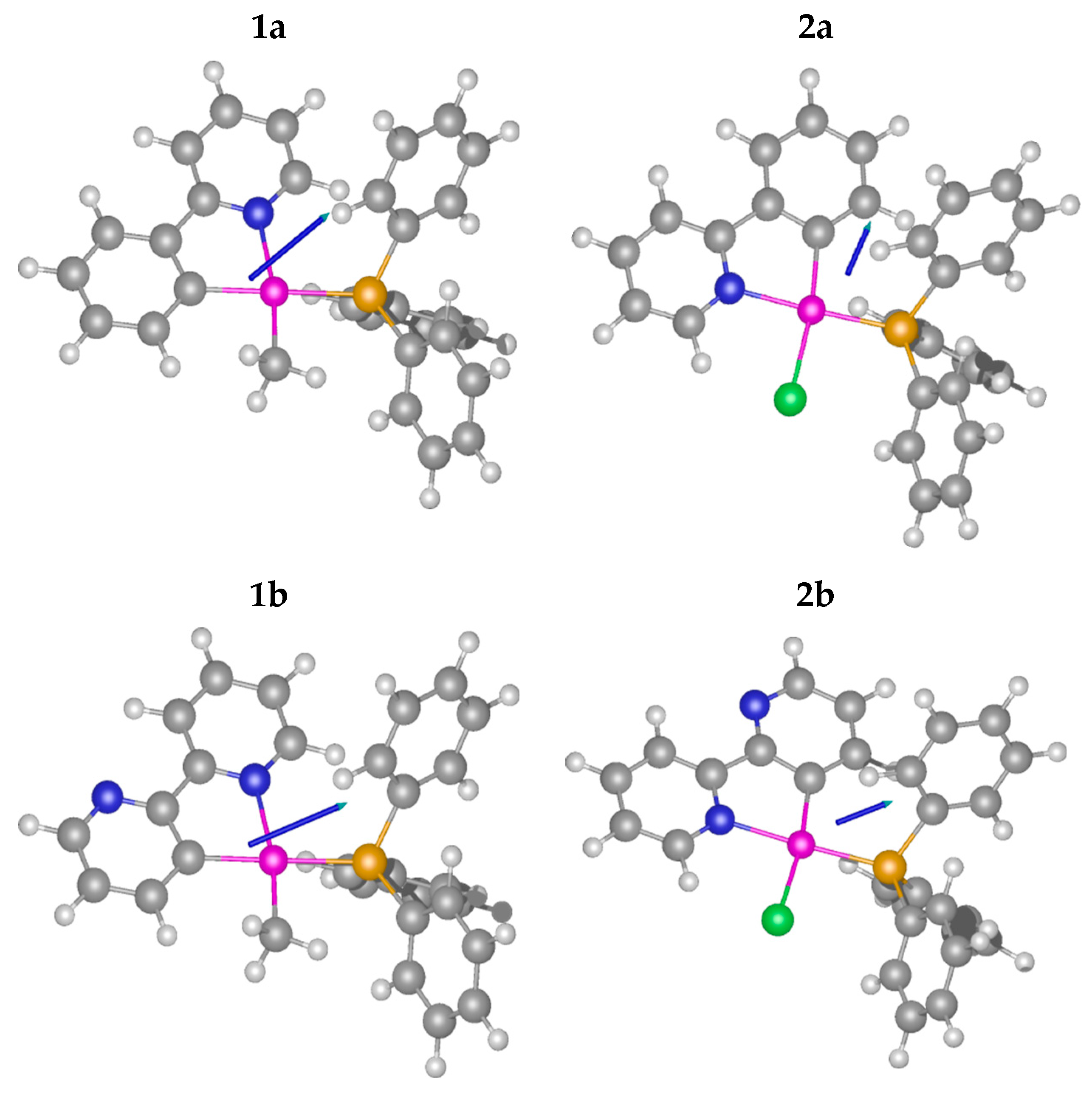
2.1. Methyl Complexes 1a and 1b
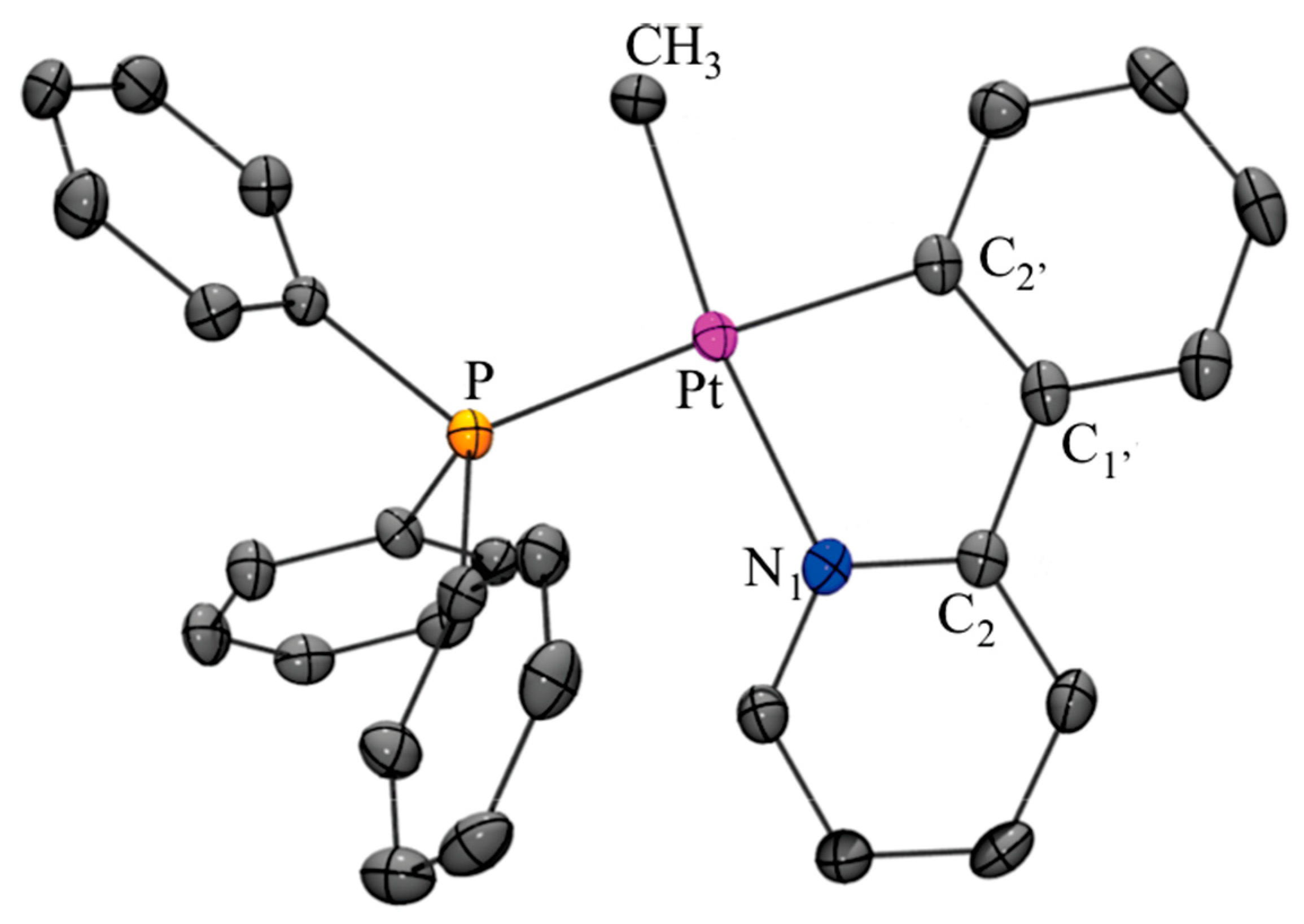
X-ray Analysis
2.2. Chlorido Complexes 2a and 2b
2.2.1. DFT Calculations
2.2.2. Electronic Spectroscopy and Electrochemical Behavior
1a and 1b
2a and 2b
3. Materials and Methods
3.1. DFT Calculations
3.2. X-ray
4. Preparations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Albrecht, M. Cyclometalation using d-block transition metals: Fundamental aspects and recent trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef]
- Dupont, J.; Pfeffer, M. Palladacycles; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Jurgens, S.; Kuhn, F.E.; Casini, A. Cyclometalated complexes of platinum and gold with biological properties: State-of-the-art and future perspectives. Curr. Med. Chem. 2018, 25, 437–461. [Google Scholar] [CrossRef]
- Abbas, S.; Din, I.-U.-D.; Raheel, A.; Tameez ud Din, A. Cyclometalated Iridium (III) complexes: Recent advances in phosphorescence bioimaging and sensing applications. Appl. Organomet. Chem. 2020, 34, e5413. [Google Scholar] [CrossRef]
- Van Koten, G. Tuning the reactivity of metals held in a rigid ligand environment. Pure Appl. Chem. 1989, 61, 1681. [Google Scholar] [CrossRef] [Green Version]
- Omae, I. Intramolecular five-membered ring compounds and their applications. Coord. Chem. Rev. 2004, 248, 995–1023. [Google Scholar] [CrossRef]
- Albrecht, M.; van Koten, G. Platinum Group Organometallics Based on “Pincer” Complexes: Sensors, Switches, and Catalysts. Angew. Chem. Int. Ed. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Omae, I. Unconventional Cyclometalation Reactions. Curr. Org. Chem. 2014, 18, 2776–2795. [Google Scholar] [CrossRef]
- Zucca, A.; Pilo, M.I. Rollover Cyclometalation as a Valuable Tool for Regioselective C–H Bond Activation and Functionalization. Molecules 2021, 26, 328. [Google Scholar] [CrossRef]
- Minghetti, G.; Stoccoro, S.; Cinellu, M.A.; Soro, B.; Zucca, A. Activation of a C-H Bond in a Pyridine RingReaction of 6-Substituted 2, 2′-Bipyridines with Methyl and Phenyl Platinum (II) Derivatives: N′, C (3)-“Rollover” Cyclometalation. Organometallics 2003, 22, 4770–4777. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Pilo, M.I.; Pischedda, S.; Sedda, M.; Stoccoro, S. Pt(II) derivatives with rollover-coordinated 6-substituted 2,20-bipyridines: Ligands with multiple personalities. Appl. Sci. 2020, 10, 6665. [Google Scholar] [CrossRef]
- Petretto, G.L.; Rourke, J.P.; Maidich, L.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G.; Clarkson, G.J.; Zucca, A. Heterobimetallic Rollover Derivatives. Organometallics 2012, 31, 2971–2977. [Google Scholar] [CrossRef]
- Maidich, L.; Zuri, G.; Stoccoro, S.; Cinellu, M.A.; Masia, M.; Zucca, A. Mesoionic complexes of platinum(II) derived from “rollover” cyclometalation: A delicate balance between Pt-C(sp3) and Pt-C(sp2) bond cleavage as a result of different reaction conditions. Organometallics 2013, 32, 438–448. [Google Scholar] [CrossRef]
- Leist, M.; Kerner, C.; Ghoochany, L.T.; Farsadpour, S.; Fizia, A.; Neu, J.P.; Schön, F.; Sun, Y.; Oelkers, B.; Lang, J.; et al. Roll-over cyclometalation: A versatile tool to enhance the catalytic activity of transition metal complexes. J. Organomet. Chem. 2018, 863, 30–43. [Google Scholar] [CrossRef]
- Crosby, S.H.; Clarkson, G.J.; Rourke, J.P. Reactions of a Platinum(II) Agostic Complex: Decyclometalation, Dicyclometalation and Solvent-Switchable Formation of a Rollover Complex. Organometallics 2011, 30, 3603–3609. [Google Scholar] [CrossRef]
- Crabtree, R.H. Creating ligands with multiple personalities. Science 2010, 330, 455–456. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Canu, L.; Petretto, G.L.; Stoccoro, S.; Cinellu, M.A.; Clarkson, G.J.; Rourke, J.P. Rollover-assisted C(sp2)-C(sp3) bond formation. Chem.-A Eur. J. 2014, 20, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Petretto, G.L.; Zucca, A.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G. Step by step palladium mediated syntheses of new 2-(pyridin-2-yl)-6-R-nicotinic acids and esters. J. Organomet. Chem. 2010, 695, 256–259. [Google Scholar] [CrossRef]
- Cuesta, L.; Soler, T.; Urriolabeitia, E.P. Cycloruthenated complexes from imine-based heterocycles: Synthesis, characterization, and reactivity toward alkynes. Chem. A Eur. J. 2012, 18, 15178–15189. [Google Scholar] [CrossRef]
- Yang, W.; Chen, J.; Huang, X.; Ding, J.; Liu, M.; Wu, H. Pd-catalyzed intramolecular aerobic oxidative C-H amination of 2-aryl-3-(arylamino)quinazolinones: Synthesis of fluorescent indazolo3,2-b.quinazolinones. Org. Lett. 2014, 16, 5418–5421. [Google Scholar] [CrossRef]
- Shibata, T.; Takayasu, S. Synthesis of Multicyclic Heterocycles Initiated by C–H Bond Activation Along with “Rollover” Using a Rh(III) Catalyst. Heteroat. Chem. 2014, 25, 379–388. [Google Scholar] [CrossRef]
- Kwak, J.; Ohk, Y.; Jung, Y.; Chang, S. Rollover Cyclometalation Pathway in Rhodium Catalysis: Dramatic NHC Effects in the C−H Bond Functionalization. J. Am. Chem. Soc. 2012, 134, 17778–17788. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Ghorai, D.; Choudhury, J. To “Rollover” or Not? Stereoelectronically Guided C–H Functionalization Pathways from Rhodium–Abnormal NHC Intermediates. ACS Omega 2018, 3, 1614–1620. [Google Scholar] [CrossRef]
- Yu, J.; Lv, W.; Cheng, G. Palladium-Catalyzed Site-Selective C-H Arylation of 2,2′-Bipyridine-6-carboxamides via a Rollover Cyclometalation Pathway. Org. Lett. 2018, 20, 4732–4735. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wen, S.; Ba, D.; Lv, W.; Chen, Y.; Cheng, G. Rhodium(III)-Catalyzed Regioselective C3−H Acylmethylation of 2,2′-Bipyridine.-6-carboxamides with Sulfoxonium Ylides. Org. Lett. 2019, 21, 6366–6369. [Google Scholar] [CrossRef]
- Alam, P.; Kaur, G.; Chakraborty, S.; Roy Choudhury, A.; Laskar, I.R. Aggregation induced phosphorescence active rollover iridium(III) complex as a multi-stimuli-responsive luminescence material. Dalton Trans. 2015, 44, 6581–6592. [Google Scholar] [CrossRef] [PubMed]
- Paziresh, S.; Babadi Aghakhanpour, R.; Rashidi, M.; Nabavizadeh, S.M. Simple tuning of the luminescence properties of the double rollover cycloplatinated(II) structure by halide ligands. New J. Chem. 2018, 42, 1337–1346. [Google Scholar] [CrossRef]
- Aghakhanpour, R.B.; Nabavizadeh, S.M.; Rashidi, M. Newly designed luminescent di- and tetra-nuclear double rollover cycloplatinated(II) complexes. J. Organomet. Chem. 2016, 819, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Abedi, A.; Amani, V.; Safari, N.; Ostad, S.N.; Notash, B. From proton transferred to cyclometalated platinum(IV) complex: Crystal structure and biological activity. J. Organomet. Chem. 2015, 799–800, 30–37. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Shahsavari, H.R.; Abedanzadeh, S.; Behchenari, B.; Hossein-Abadi, M.; Faghih, Z.; Hassan Beyzavi, M. Cycloplatinated(II) complexes bearing 1,1′-bis(diphenylphosphino)ferrocene ligand: Biological evaluation and molecular docking studies. New J. Chem. 2018, 42, 2385–2392. [Google Scholar] [CrossRef]
- Akbarzadeh, S.; Ebrahimi, F.; Faghih, Z.; Movahed, A.; Faghih, Z. Cytotoxic Effect Two Novel Platinum Breast Cancer: An in Vitro Study. Asian Pac. J. Cancer Biol. 2018, 3, 11–14. [Google Scholar] [CrossRef]
- Babak, M.V.; Pfaffeneder-Kmen, M.; Meier-Menches, S.M.; Legina, M.S.; Theiner, S.; Licona, C.; Orvain, C.; Hejl, M.; Hanif, M.; Jakupec, M.A.; et al. Rollover Cyclometalated Bipyridine Platinum Complexes as Potent Anticancer Agents: Impact of the Ancillary Ligands on the Mode of Action. Inorg. Chem. 2018, 57, 2851–2864. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Niazi, M.; Shahmohammadi Beni, M.; Mohammadi, S.; Faghih, Z.; Faghih, Z.; Shahsavari, H.R. Synthesis, Biological Evaluation, and Molecular Docking Studies on the DNA Binding Interactions of Platinum(II) Rollover Complexes Containing Phosphorus Donor Ligands. Chem. Med. Chem. 2017, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Aghakhanpour, R.B.; Nabavizadeh, S.M.; Mohammadi, L.; Jahromi, S.A.; Rashidi, M. A kinetic approach to carbon-iodide bond activation by rollover cycloplatinated(II) complexes containing monodentate phosphine ligands. J. Organomet. Chem. 2015, 781, 47–52. [Google Scholar] [CrossRef]
- Edwards, G.L.; Black, D.S.; Deacon, G.B.; Wakelin, L.P. In vitro and in vivo studies of neutral cyclometallated complexes against murine leukæmias. Can. J. Chem. 2005, 83, 980–989. [Google Scholar] [CrossRef]
- Samouei, H.; Rashidi, M.; Heinemann, F.W. Cyclometalated platinum(II) complexes containing monodentate phosphines: Antiproliferative study. J. Iran. Chem. Soc. 2014, 11, 1207–1216. [Google Scholar] [CrossRef]
- Shahsavari, H.R.; Babadi Aghakhanpour, R.; Babaghasabha, M.; Golbon Haghighi, M.; Nabavizadeh, S.M.; Notash, B. Photophysical properties of a series of cycloplatinated(II) complexes featuring allyldiphenylphosphane. New J. Chem. 2017, 41, 3798–3810. [Google Scholar] [CrossRef]
- Jamali, S.; Nabavizadeh, S.M.; Rashidi, M. 1,1′ Bis(diphenylphosphino) ferrocene as Spacer Ligand: Kinetics and Mechanism of MeI Oxidative Addition. Inorg. Chem. 2008, 47, 5441–5452. [Google Scholar] [CrossRef]
- Rourke, J.; Maidich, L.; Clarkson, G.; University of Sassari, Sassari, Italy. Private Communication, 2017.
- Zucca, A.; Petretto, G.L.; Stoccoro, S.; Agostina, M.; Manassero, M.; Manassero, C.; Minghetti, G.; Cinellu, M.A. Cyclometalation of 2,2′-Bipyridine. Mono-and Dinuclear C, N Platinum (II) Derivatives. Organometallics 2009, 28, 2150–2159. [Google Scholar] [CrossRef]
- Münzenberg, R.; Rademacher, P.; Boese, R. Chiral platinum (II) complexes with phosphorus derivatives of the amino acid L-proline. NMR spectroscopic and X-ray structure investigations of the cis influence of tertiary phosphorus ligands. J. Mol. Struct. 1998, 444, 77–90. [Google Scholar] [CrossRef]
- Still, B.M.; Kumar, P.G.A.; Aldrich-Wright, J.R.; Price, W.S. 195Pt NMR—Theory and application. Chem. Soc. Rev. 2007, 36, 665–686. [Google Scholar] [CrossRef]
- Maidich, L.; Dettori, G.; Stoccoro, S.; Cinellu, M.A.; Rourke, J.P.; Zucca, A. Electronic and steric effects in rollover C-H bond activation. Organometallics 2015, 34, 817–828. [Google Scholar] [CrossRef]
- Owen, J.S.; Labinger, J.A.; Bercaw, J.E. Pyridinium-Derived N-Heterocyclic Carbene Complexes of Platinum: Synthesis, Structure and Ligand Substitution Kinetics. JACS 2004, 126, 8247–8255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maheshwari, V.; Carlone, M.; Fronczek, F.R.; Marzilli, L.G. Ligand and coordination-plane distortions in platinum(II) complexes of isomers of dimethyl-2,2′-bipyridine. Acta Crystallogr. Sect. B Struct. Sci. 2007, 63, 603–611. [Google Scholar] [CrossRef]
- Hazell, A. Is bipyridine planar in metal complexes? Polyhedron 2004, 23, 2081–2083. [Google Scholar] [CrossRef]
- Geremia, S.; Randaccio, L.; Mestroni, G.; Milani, B. Bow-step and twist conformations and stacking interactions in palladium bipyridine and phenanthroline complexes. J. Chem. Soc. Dalt. Trans. 1992, 2117–2118. [Google Scholar] [CrossRef]
- Pérez, J.; García, L.; Pérez, E.; Serrano, J.L.; Martínez, J.F.; Sánchez, G.; López, G.; Espinosa, A.; Liu, M.; Sanz, F. Solid state conformational and theoretical study of complexes containing the (CxN)Pd moiety (CxN = 2-(phenylazo)phenyl-C,N and its derivatives). New J. Chem. 2003, 27, 1490–1496. [Google Scholar] [CrossRef]
- Solomatina, A.I.; Chelushkin, P.S.; Krupenya, D.V.; Podkorytov, I.S.; Artamonova, T.O.; Sizov, V.V.; Melnikov, A.S.; Gurzhiy, V.V.; Koshel, E.I.; Shcheslavskiy, V.I.; et al. Coordination to Imidazole Ring Switches on Phosphorescence of Platinum Cyclometalated Complexes: The Route to Selective Labeling of Peptides and Proteins via Histidine Residues. Bioconjug. Chem. 2017, 28, 426–437. [Google Scholar] [CrossRef]
- Sivchik, V.; Sarker, R.K.; Liu, Z.Y.; Chung, K.Y.; Grachova, E.V.; Karttunen, A.J.; Chou, P.T.; Koshevoy, I.O. Improvement of the photophysical performance of platinum-cyclometalated complexes in halogen-bonded adducts. Chem.-A Eur. J. 2018, 24, 11475–11484. [Google Scholar] [CrossRef]
- Niazi, M.; Shahsavari, H.R.; Haghighi, M.G.; Halvagar, M.R.; Hatami, S.; Notash, B. Carbon-sulfur bond reductive coupling from a platinum(II) thiolate complex. RSC Adv. 2016, 6, 95073–95084. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson Education Limited: Harlow, UK, 2018. [Google Scholar]
- Sanna, G. Platinum complexes with N_N_C ligands. Syntheses, electrochemical and spectroscopic characterisations of platinum(II) and relevant electroreduced species. Inorg. Chim. Acta 2000, 305, 189–205. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Carta, V.; Petretto, G.P.; Stoccoro, S.; Cinellu, M.A.; Pilo, M.I.; Clarkson, G.J. Cyclometalated complexes of platinum(II) with 2-vinylpyridine. Eur. J. Inorg. Chem. 2014, 13, 2278–2287. [Google Scholar] [CrossRef]
- Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Scientific and Technical: Harlow, UK, 1989.
- Eaborn, C.; Kundu, K.; Pidcock, A. Synthesis of platinum(II) alkyl and aryl complexes from K2PtCl4. and tetraorganotin compounds in dimethyl sulphoxide. J. Chem. Soc. Dalton Trans. 1981, 4, 933–938. [Google Scholar] [CrossRef]
- Romeo, R.; Scolaro, L.M. (2,2′: 6′,2″-Terpyridine) Methylplatinum(II) Chloride and (1,10-Phenanthroline) Methylchloroplatinum(II). Inorg. Synth. 1998, 32, 153. [Google Scholar]
- Baquero, E.A.; Rodríguez-Zúñiga, A.; Flores, J.C.; Temprado, M.; de Jesús, E. Revisiting the synthesis of trans-Pt(dmso)2ClMe. and cis-Pt(dmso)2Me2: Experimental and DFT studies. J. Organomet. Chem. 2019, 896, 108–112. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Y.; Gao, J.; Wang, D.; Yu, G.; Heeger, A.J. Electrochemical properties of luminescent polymers and polymer light-emitting electrochemical cells. Synth. Met. 1999, 99, 243–248. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum. Phys. Rev. Lett. 1997, 78, 1396–1396. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Chang, C.; Pelissier, M.; Durand, P. Regular Two-Component Pauli-Like Effective Hamiltonians in Dirac Theory. Phys. Scr. 1986, 34, 394–404. [Google Scholar] [CrossRef]
- Heully, J.L.; Lindgren, I.; Lindroth, E.; Lundqvist, S.; Martensson-Pendrill, A.M. Diagonalisation of the Dirac Hamiltonian as a basis for a relativistic many-body procedure. J. Phys. B At. Mol. Phys. 1986, 19, 2799–2815. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.J. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Alex, A. Granovsky, Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 1 October 2022).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput.Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular Interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef] [Green Version]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Lu, T. Generalized Charge Decomposition Analysis (GCDA) Method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.J. Multiwfn: A multifunctional wavefunction analyzer. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 3, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Available online: http://www.povray.org (accessed on 1 October 2022).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
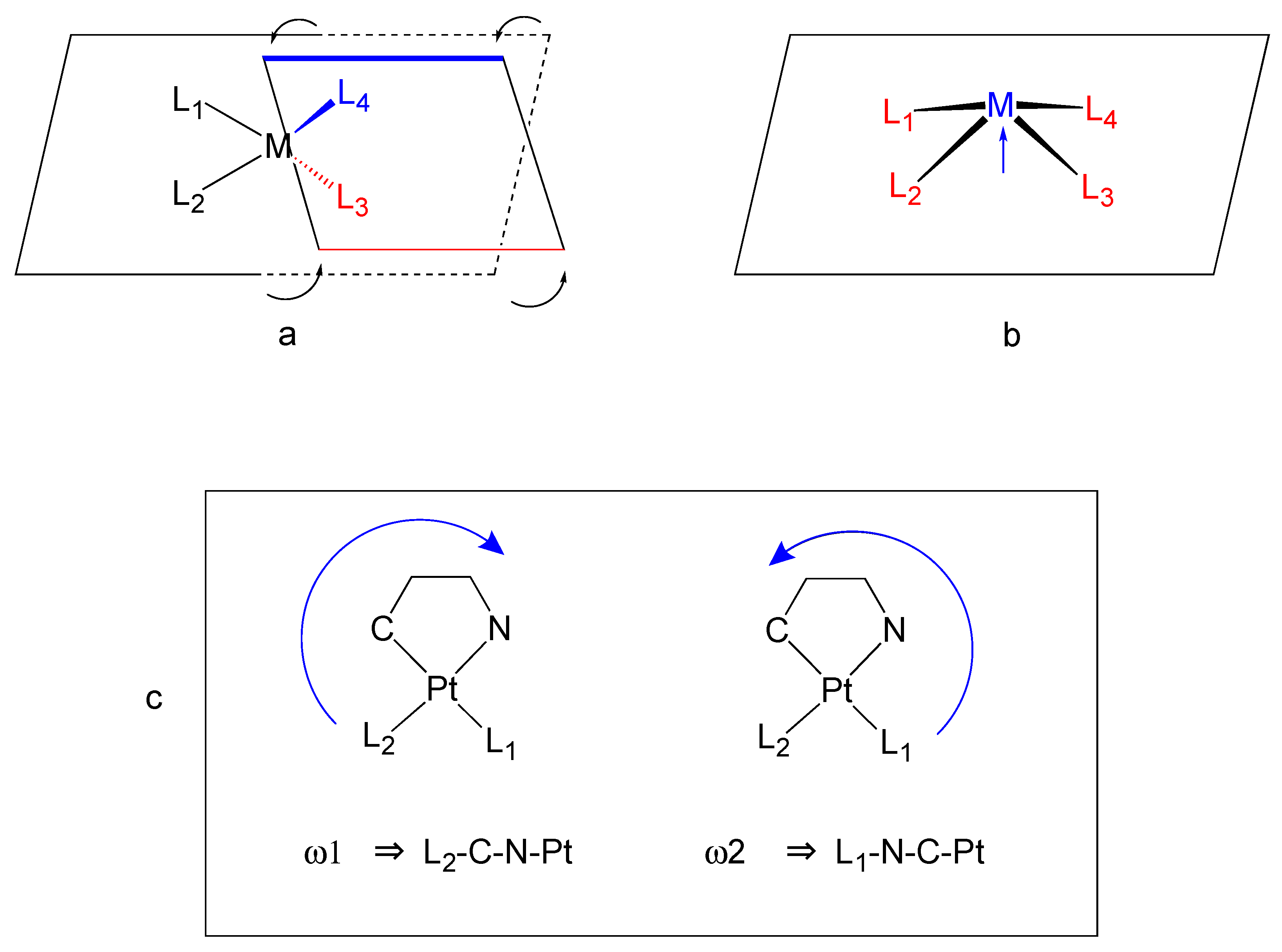
| 1a′ | 1a″ | 1b | 2a′ | 2a″ | 2a‴ | 2a′′′′ | 2b(mol1) | 2b′(mol2) | |
|---|---|---|---|---|---|---|---|---|---|
| CCDC | 1546694 | 1457812 | 1505457 | 1505459 | |||||
| ω1 | −0.54 (9) | −1.5 (3) | +4.43 (8) | −1.34 (3) | −1.5 (1) | −3.8 (1) | +1.32 (6) | −1.09 (4) | −4.89 (4) |
| ω2 | −3.12 (7) | −2.5 (2) | +4.01 (6) | +1.00 (4) | +0.7 (2) | −2.9 (1) | −1.04 (6) | −2.35 (5) | −2.57 (5) |
| Pl1/Pl2 d.a. | 4.55 | 4.13 | 8.48 | 6.16 | 5.86 | 6.43 | 4.57 | 5.04 | 2.36 |
| d.a. from Pl3 | |||||||||
| Pl4(pyN) | 9.91 | 9.90 | 10.82 | 0.73 | 0.71 | 0.83 | 6.91 | 4.02 | 7.84 |
| Pl5(PyC) | 7.91 | 7.41 | 10.29 | 5.44 | 5.25 | 5.62 | 4.43 | 5.98 | 7.72 |
| Distance from Pl1 a | |||||||||
| C1 or Cl | +0.028 | +0.076 | −0.227 | +0.038 | +0.056 | −0.165 | −0.058 | −0.121 | −0.143 |
| P1 | −0.180 | −0.143 | +0.233 | +0.083 | +0.073 | +0.209 | −0.072 | +0.069 | +0.264 |
| d.a. between Pl4/Pl5 (PyN/PyC) | 8.79 | 6.60 | 6.63 | 6.16 | 5.86 | 6.43 | 4.57 | 5.04 | 2.36 |
| Complex | Gas-Phase | CH2Cl2 | ||
|---|---|---|---|---|
| ΔH | ΔG | ΔH | ΔG | |
| 1a | 0.31 | 1.33 | 2.13 | 1.72 |
| 1b | 1.34 | 2.63 | 3.49 | 4.34 |
| 2a | 8.60 | 7.62 | 8.58 | 9.44 |
| 2b | 8.09 | 7.11 | 8.96 | 9.16 |
| Correct Isomer P-trans-C for 1a-b and P-trans-N for 2a-b | Inverted Isomer P-trans-N for 1a-b and P-trans-C for 2a-b | |
|---|---|---|
| Pt-P dihedral scan | ||
| [Pt(bpy-H)(Cl)(PPh3)] | 16.6 | 11.1 |
| [Pt(ppy-H)(Cl)(PPh3)] | 17.1 | 15.3 |
| [Pt(bpy-H)(Me)(PPh3)] | 11.7 | 14.8 |
| [Pt(ppy-H)(Me)(PPh3)] | 12.3 | 15.4 |
| Pt-CH3 dihedral scan | ||
| Phenyl rings of PPh3 | above/below bpy | above/below bpy |
| [Pt(bpy-H)(Me)(PPh3)] | 5.0 | 5.1 |
| [Pt(ppy-H)(Me)(PPh3)] | 5.8 | 5.6 |
| ECDA Results | DFT Interaction Energies | ||||||
|---|---|---|---|---|---|---|---|
| C^N ligand | PPh3 ligand | Me | Cl | bpy-H | ppy-H | PPh3 | |
| 1a | −0.9335 | −0.3237 | −1075.55 | - | - | −1236.82 | −310.10 |
| 1b | −0.9304 | −0.3330 | −1093.57 | - | −1213.46 | - | −317.76 |
| 2a | −1.0335 | −0.4565 | - | −721.78 | - | −1417.02 | −422.58 |
| 2b | −1.0288 | −0.4652 | - | −735.67 | −1390.62 | - | −428.59 |
| Complex | WE = Pt CH2Cl2/TEAPF6 0.1 M | |||||||
|---|---|---|---|---|---|---|---|---|
| Eox (V) | HOMO (eV) | HOMO (eV) Theor | λ (nm) (ε, L mol−1 cm−1) | Eopt (eV) | Etheor (eV) | LUMO (eV) | LUMO (eV) Theor | |
| 1a | 0.83 | −5.65 | −5.77 (gas) −6.16 (CH2Cl2) | 268 (2.0 × 104) 328 (3.4 × 103) 358 (3.6 × 103) | 3.07 | 4.33 | −2.54 | −1.44ca (gas) −1.63 (CH2Cl2) |
| 2a | 1.01 | −5.96 | −5.68 (gas) −6.02 (CH2Cl2) | 286 (1.2 × 104) 378 (2.0 × 103) | 2.87 | 4.01 | −2.86 | −1.67 (gas) −1.76 (CH2Cl2) |
| 1b | 0.95 | −5.67 | −5.89 (gas) −6.28 (CH2Cl2) | 254 (2.0 × 104) 313 (7.0 × 103) 352 (1.5 × 103) | 3.10 | 4.38 | −2.28 | −1.51 (gas) −1.72 (CH2Cl2) |
| 2b | 1.17 | −6.00 | −5.82 (gas) −6.14 (CH2Cl2) | 258 (1.6 × 104) 300(S),372 (2.6 × 103) | 3.00 | 4.08 | −2.46 | −1.74 (gas) −1.86 (CH2Cl2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maidich, L.; Pilo, M.I.; Rourke, J.P.; Clarkson, G.J.; Canu, P.; Stoccoro, S.; Zucca, A. Classical vs. Non-Classical Cyclometalated Pt(II) Complexes. Molecules 2022, 27, 7249. https://doi.org/10.3390/molecules27217249
Maidich L, Pilo MI, Rourke JP, Clarkson GJ, Canu P, Stoccoro S, Zucca A. Classical vs. Non-Classical Cyclometalated Pt(II) Complexes. Molecules. 2022; 27(21):7249. https://doi.org/10.3390/molecules27217249
Chicago/Turabian StyleMaidich, Luca, Maria I. Pilo, Jonathan P. Rourke, Guy J. Clarkson, Patrizia Canu, Sergio Stoccoro, and Antonio Zucca. 2022. "Classical vs. Non-Classical Cyclometalated Pt(II) Complexes" Molecules 27, no. 21: 7249. https://doi.org/10.3390/molecules27217249