
Crosstalk between Depression and Breast Cancer via Hepatic Epoxide Metabolism: A Central Comorbidity Mechanism
 , ,
, , 
Abstract
:1. Introduction
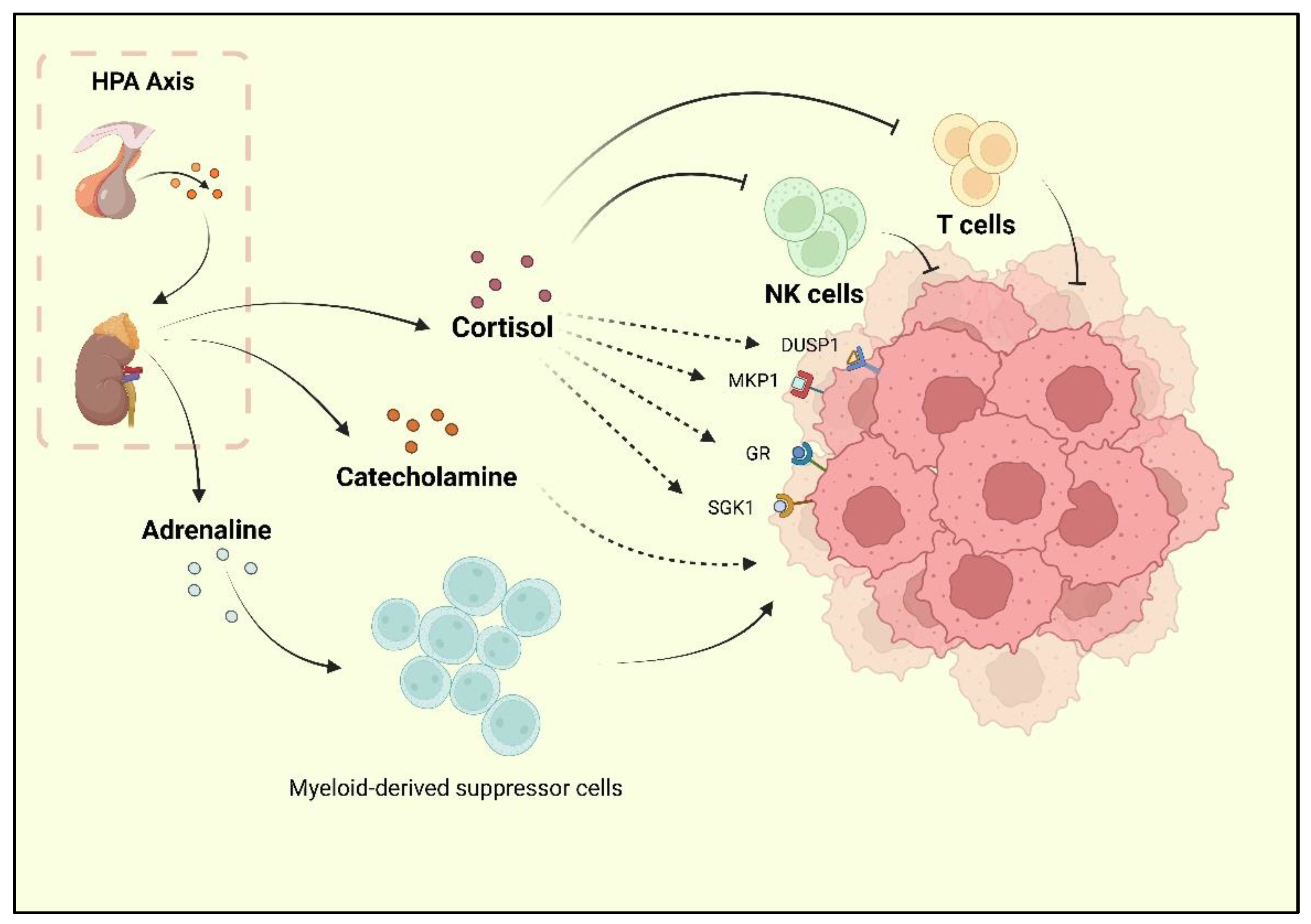
2. Depression Is an Important Risk Factor and Comorbidity of BC
3. Specific Changes in Epoxide Metabolism in Individuals with Depression
4. Different Status of sEH Mediates Epoxide Metabolism in BC
5. sEH-Mediated Epoxide Metabolism Is Involved in the TME of BC
6. Depression-Associated sEH Promotes Liver Dysfunction and Breast Cancer
7. Prospective Studies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Msc, M.L.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chan, P.S.; Lok, V.; Chen, X.; Ding, H.; Jin, Y.; Yuan, J.; Lao, X.-Q.; Zheng, Z.-J.; Wong, M.C. Global incidence and mortality of breast cancer: A trend analysis. Aging 2021, 13, 5748–5803. [Google Scholar] [CrossRef] [PubMed]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Apaya, M.K.; Shiau, J.-Y.; Liao, G.-S.; Liang, Y.-J.; Chen, C.-W.; Yang, H.-C.; Chu, C.-H.; Yu, J.-C.; Shyur, L.-F. Integrated omics-based pathway analyses uncover CYP epoxygenase-associated networks as theranostic targets for metastatic triple negative breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Puigpinós-Riera, R.; Graells-Sans, A.; Serral, G.; Continente, X.; Bargalló, X.; Domènech, M.; Espinosa-Bravo, M.; Grau, J.; Macià, F.; Manzanera, R.; et al. Anxiety and depression in women with breast cancer: Social and clinical determinants and influence of the social network and social support (DAMA cohort). Cancer Epidemiol. 2018, 55, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Kissane, D.W.; Grabsch, B.; Love, A.; Clarke, D.M.; Bloch, S.; Smith, G.C. Psychiatric Disorder in Women with Early Stage and Advanced Breast Cancer: A Comparative Analysis. Aust. N. Z. J. Psychiatry 2004, 38, 320–326. [Google Scholar] [CrossRef]
- Pelletier, G.; Verhoef, M.J.; Khatri, N.; Hagen, N. Quality of Life in Brain Tumor Patients: The Relative Contributions of Depression, Fatigue, Emotional Distress, and Existential Issues. J. Neuro-Oncol. 2002, 57, 41–49. [Google Scholar] [CrossRef]
- Desai, M.; Bruce, M.L.; Kasl, S.V. The Effects of Major Depression and Phobia on Stage at Diagnosis of Breast Cancer. Int. J. Psychiatry Med. 1999, 29, 29–45. [Google Scholar] [CrossRef]
- Satin, J.R.; Linden, W.; Phillips, M.J. Depression as a predictor of disease progression and mortality in cancer patients. Cancer 2009, 115, 5349–5361. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Yoshikawa, E.; Matsuoka, Y.; Sugawara, Y.; Nakano, T.; Akechi, T.; Wada, N.; Imoto, S.; Murakami, K.; Uchitomi, Y.; et al. Smaller regional volumes of brain gray and white matter demonstrated in breast cancer survivors exposed to adjuvant chemotherapy. Cancer 2006, 109, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Han, R.; Yang, Y.; Mayer-Pröschel, M.; Noble, M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol. 2006, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Janelsins, M.C.; Roscoe, J.A.; Berg, M.J.; Thompson, B.D.; Gallagher, M.J.; Morrow, G.R.; Heckler, C.E.; Jean-Pierre, P.; Opanashuk, L.A.; Gross, R.A. IGF-1 Partially Restores Chemotherapy-Induced Reductions in Neural Cell Proliferation in Adult C57BL/6 Mice. Cancer Investig. 2009, 28, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Christie, L.-A.; Acharya, M.M.; Parihar, V.K.; Nguyen, A.; Martirosian, V.; Limoli, C.L. Impaired Cognitive Function and Hippocampal Neurogenesis following Cancer Chemotherapy. Clin. Cancer Res. 2012, 18, 1954–1965. [Google Scholar] [CrossRef] [Green Version]
- Chapman, B.; Swainston, J.; Grunfeld, E.A.; Derakshan, N. COVID-19 Outbreak Effects on Job Security and Emotional Functioning Amongst Women Living with Breast Cancer. Front. Psychol. 2020, 11, 582014. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ganesan, K.; Liu, X.; Ye, Q.; Cheung, Y.; Liu, D.; Zhong, S.; Chen, J. Prognostic Value of Negative Emotions on the Incidence of Breast Cancer: A Systematic Review and Meta-Analysis of 129,621 Patients with Breast Cancer. Cancers 2022, 14, 475. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, B.; Hyphantis, T.N.; Valpione, S.; Perini, G.; Maes, M.; Morris, G.; Kubera, M.; Köhler, C.A.; Fernandes, B.S.; Stubbs, B.; et al. Depression in cancer: The many biobehavioral pathways driving tumor progression. Cancer Treat. Rev. 2016, 52, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Tejada, J.; Labaka, A.; Vegas, O.; Larraioz, A.; Pescador, A.; Arregi, A. Anxiety and depression after breast cancer: The predictive role of monoamine levels. Eur. J. Oncol. Nurs. 2021, 52, 101953. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.W.; Morisseau, C.; Hammock, B.D. Epoxide hydrolases: Their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005, 44, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C. Role of epoxide hydrolases in lipid metabolism. Biochime 2013, 95, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K. Role of Soluble Epoxide Hydrolase in Metabolism of PUFAs in Psychiatric and Neurological Disorders. Front. Pharmacol. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markaverich, B.M.; Crowley, J.R.; Alejandro, M.A.; Shoulars, K.; Casajuna, N.; Mani, S.; Reyna, A.; Sharp, J. Leukotoxin Diols from Ground Corncob Bedding Disrupt Estrous Cyclicity in Rats and Stimulate MCF-7 Breast Cancer Cell Proliferation. Environ. Health Perspect. 2005, 113, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Markaverich, B.; Mani, S.; Alejandro, M.A.; Mitchell, A.; Markaverich, D.; Brown, T.; Velez-Trippe, C.; Murchison, C.; O’Malley, B.; Faith, R. A novel endocrine-disrupting agent in corn with mitogenic activity in human breast and prostatic cancer cells. Environ. Health Perspect. 2002, 110, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Fagundes, C.P.; Glaser, R.; Hwang, B.S.; Malarkey, W.B.; Kiecolt-Glaser, J.K. Depressive symptoms enhance stress-induced inflammatory responses. Brain, Behav. Immun. 2012, 31, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.-S.; Tseng, H.-Y.; Chen, Y.-A.; Shen, P.-C.; Al Haq, A.T.; Chen, L.-M.; Tung, Y.-C.; Hsu, H.-L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, J.; Liu, L.; Wang, F.; Ma, Z.; Gao, D.; Zhang, Q.; Yu, Z. The status and correlates of depression and anxiety among breast-cancer survivors in Eastern China: A population-based, cross-sectional case–control study. BMC Public Health 2014, 14, 326. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.W.-L.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: The impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol. Immunother. 2014, 63, 1115–1128. [Google Scholar] [CrossRef]
- Gosain, R.; Gage-Bouchard, E.; Ambrosone, C.; Repasky, E.; Gandhi, S. Stress reduction strategies in breast cancer: Review of pharmacologic and non-pharmacologic based strategies. Semin. Immunopathol. 2020, 42, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Kach, J.; Conzen, S.D.; Szmulewitz, R.Z. Targeting the glucocorticoid receptor in breast and prostate cancers. Sci. Transl. Med. 2015, 7, 305ps19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Pompe, G.; Antoni, M.H.; Mulder, C.L.; Heijnen, C.; Goodkin, K.; De Graeff, A.; Garssen, B.; De Vries, M.J. Psychoneuroimmunology and the course of breast cancer: An overview the impact of psychosocial factors on progression of breast cancer through immune and endocrine mechanisms. Psycho-Oncology 1994, 3, 271–288. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Pace, T.W.; Mletzko, T.C.; Alagbe, O.; Musselman, D.L.; Nemeroff, C.B.; Miller, A.H.; Heim, C.M. Increased Stress-Induced Inflammatory Responses in Male Patients with Major Depression and Increased Early Life Stress. Am. J. Psychiatry 2006, 163, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity Promotes Breast Cancer by CCL2-Mediated Macrophage Recruitment and Angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santander, A.M.; Lopez-Ocejo, O.; Casas, O.; Agostini, T.; Sanchez, L.; Lamas-Basulto, E.; Carrio, R.; Cleary, M.P.; Gonzalez-Perez, R.R.; Torroella-Kouri, M. Paracrine Interactions between Adipocytes and Tumor Cells Recruit and Modify Macrophages to the Mammary Tumor Microenvironment: The Role of Obesity and Inflammation in Breast Adipose Tissue. Cancers 2015, 7, 143–178. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Egbrink, M.G.A.O.; Wagstaff, J.; Griffioen, A.W. Monocyte/macrophage infiltration in tumors: Modulators of angiogenesis. J. Leukoc. Biol. 2006, 80, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Junjeong, C.; Jones, G.; Haerin, J.; Seung, K.J. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2017, 33, 133–145. [Google Scholar] [CrossRef]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhallak, I.; Wolter, K.G.; Munoz, A.C.; Simmen, F.A.; Ward, R.J.; Petty, S.A.; Li, L.-X.; Simmen, R.C. Breast adipose regulation of premenopausal breast epithelial phenotype involves interleukin 10. J. Mol. Endocrinol. 2021, 67, 173–188. [Google Scholar] [CrossRef]
- Qiu, X.; Zhao, T.; Luo, R.; Qiu, R.; Li, Z. Tumor-Associated Macrophages: Key Players in Triple-Negative Breast Cancer. Front. Oncol. 2022, 12, 772615. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Workman, C.J.; Beyaz, S.; LoCascio, S.; Zhao, G.; Vignali, D.A.A.; Sykes, M. LAG-3, TGF-β, and cell-intrinsic PD-1 inhibitory pathways contribute to CD8 but not CD4 T-cell tolerance induced by allogeneic BMT with anti-CD40L. Blood 2011, 117, 5532–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, P.-Y.; Wang, S.-M.; Chen, P.-M.; Tang, F.-Y.; Chiang, E.-P.I. Expression of MTDH and IL-10 Is an Independent Predictor of Worse Prognosis in ER-Negative or PR-Negative Breast Cancer Patients. J. Clin. Med. 2020, 9, 3153. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-Y.; Chien, W.-C.; Liao, J.-M.; Tsai, C.-W.; Chang, W.-S.; Su, C.-H.; Hsu, S.-W.; Wang, H.-C.; Bau, D.-T. Contribution of Interleukin-10 Genotype to Triple Negative Breast Cancer Risk. Anticancer Res. 2021, 41, 2451–2457. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Derry, H.M.; Fagundes, C.P. Inflammation: Depression Fans the Flames and Feasts on the Heat. Am. J. Psychiatry 2015, 172, 1075–1091. [Google Scholar] [CrossRef]
- Lamers, F.; Milaneschi, Y.; Smit, J.H.; Schoevers, R.A.; Wittenberg, G.; Penninx, B.W. Longitudinal Association Between Depression and Inflammatory Markers: Results from the Netherlands Study of Depression and Anxiety. Biol. Psychiatry 2019, 85, 829–837. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Uzzan, S.; Azab, A. Anti-TNF-α Compounds as a Treatment for Depression. Molecules 2021, 26, 2368. [Google Scholar] [CrossRef] [PubMed]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.; Hardesty, J.; Zirnheld, K.; McClain, C.; Warner, D.; Kirpich, I. Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology 2020, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, J.; Proschak, E. Phosphatase activity of soluble epoxide hydrolase. Prostaglandins Other Lipid Mediat. 2017, 133, 88–92. [Google Scholar] [CrossRef]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, 1206. [Google Scholar] [CrossRef]
- Domingues, M.F.; Callai-Silva, N.; Piovesan, A.R.; Carlini, C.R. Soluble Epoxide Hydrolase and Brain Cholesterol Metabolism. Front. Mol. Neurosci. 2020, 12, 325. [Google Scholar] [CrossRef]
- Wu, Q.; Cai, H.; Song, J.; Chang, Q. The effects of sEH inhibitor on depression-like behavior and neurogenesis in male mice. J. Neurosci. Res. 2017, 95, 2483–2492. [Google Scholar] [CrossRef]
- Wada, M.; DeLong, C.J.; Hong, Y.H.; Rieke, C.J.; Song, I.; Sidhu, R.S.; Yuan, C.; Warnock, M.; Schmaier, A.H.; Yokoyama, C.; et al. Enzymes and Receptors of Prostaglandin Pathways with Arachidonic Acid-derived Versus Eicosapentaenoic Acid-derived Substrates and Products *. J. Biol. Chem. 2007, 282, 22254–22266. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Spector, A.A.; Kim, H.-Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swardfager, W.; Hennebelle, M.; Yu, D.; Hammock, B.; Levitt, A.; Hashimoto, K.; Taha, A. Metabolic/inflammatory/vascular comorbidity in psychiatric disorders; soluble epoxide hydrolase (sEH) as a possible new target. Neurosci. Biobehav. Rev. 2018, 87, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.-H.; Wu, Z.; Dong, J.-H.; Zeng, Y.-N.; Xiong, W.-C.; Liu, C.; Wang, M.-Y.; Zhu, M.-Z.; Chen, W.-J.; Zhang, Y.; et al. Liver Soluble Epoxide Hydrolase Regulates Behavioral and Cellular Effects of Chronic Stress. Cell Rep. 2019, 29, 3223–3234.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Iglesias, N.; Nadjar, A.; Sierra, A.; Valero, J. Susceptibility of Female Mice to the Dietary Omega-3/Omega-6 Fatty-Acid Ratio: Effects on Adult Hippocampal Neurogenesis and Glia. Int. J. Mol. Sci. 2022, 23, 3399. [Google Scholar] [CrossRef]
- Daskalopoulos, E.P.; Malliou, F.; Rentesi, G.; Marselos, M.; Lang, M.A.; Konstandi, M. Stress is a critical player in CYP3A, CYP2C, and CYP2D regulation: Role of adrenergic receptor signaling pathways. Am. J. Physiol. Metab. 2012, 303, E40–E54. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, Y.; Chang, L.; Hammock, B.D.; Hashimoto, K. Increased expression of soluble epoxide hydrolase in the brain and liver from patients with major psychiatric disorders: A role of brain—Liver axis. J. Affect. Disord. 2020, 270, 131–134. [Google Scholar] [CrossRef]
- Panigrahy, D.; Kaipainen, A.; Greene, E.R.; Huang, S. Cytochrome P450-derived eicosanoids: The neglected pathway in cancer. Cancer Metastasis Rev. 2010, 29, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Panigrahy, D.; Edin, M.L.; Lee, C.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnés, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yao, J.F.; Deng, X.F.; Zheng, X.D.; Jia, M.; Wang, Y.Q.; Huang, Y.; Zhu, J.H. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin αvβ3 and activating FAK/PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2018, 37, 1–11. [Google Scholar] [CrossRef]
- Zhu, P.; Peck, B.; Licea-Perez, H.; Callahan, J.F.; Booth-Genthe, C. Development of a semi-automated LC/MS/MS method for the simultaneous quantitation of 14,15-epoxyeicosatrienoic acid, 14,15-dihydroxyeicosatrienoic acid, leukotoxin and leukotoxin diol in human plasma as biomarkers of soluble epoxide hydrolase activity in vivo. J. Chromatogr. B 2011, 879, 2487–2493. [Google Scholar] [CrossRef]
- Hildreth, K.; Kodani, S.D.; Hammock, B.D.; Zhao, L. Cytochrome P450-derived linoleic acid metabolites EpOMEs and DiHOMEs: A review of recent studies. J. Nutr. Biochem. 2020, 86, 108484. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef]
- Rody, A.; Karn, T.; Liedtke, C.; Pusztai, L.; Ruckhaeberle, E.; Hanker, L.; Gaetje, R.; Solbach, C.; Ahr, A.; Metzler, D.; et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011, 13, R97. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Wang, Y.; Xiao, C.; Johnsen, H.; Naume, B.; Samaha, R.R.; Børresen-Dale, A.-L. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: Gene expression analyses across three different platforms. BMC Genom. 2006, 7, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquet, E.; Hallett, M.T. Absolute Assignment of Breast Cancer Intrinsic Molecular Subtype. JNCI J. Natl. Cancer Inst. 2014, 107, 357. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2010, 24, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Phuong, N.T.T.; Kim, J.W.; Kim, J.-A.; Jeon, J.S.; Lee, J.-Y.; Xu, W.J.; Yang, J.W.; Kim, S.K.; Kang, K.W. Role of the CYP3A4-mediated 11,12-epoxyeicosatrienoic acid pathway in the development of tamoxifen-resistant breast cancer. Oncotarget 2017, 8, 71054–71069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Hoy, A.J.; Balaban, S.; Saunders, D. Adipocyte–Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and Cancer: Local and Systemic Mechanisms. Annu. Rev. Med. 2015, 66, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, S. Mechanistic insights of adipocyte metabolism in regulating breast cancer progression. Pharmacol. Res. 2020, 155, 104741. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Yang, P.-B.; Hou, P.-P.; Liu, F.-Y.; Hong, W.-B.; Chen, H.-Z.; Sun, X.-Y.; Li, P.; Zhang, Y.; Ju, C.-Y.; Luo, L.-J.; et al. Blocking PPARγ interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27412–27422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dang, H.; Li, D.; Pang, W.; Hammock, B.D.; Zhu, Y. Inhibition of Soluble Epoxide Hydrolase Attenuates High-Fat-Diet–Induced Hepatic Steatosis by Reduced Systemic Inflammatory Status in Mice. PLoS ONE 2012, 7, e39165. [Google Scholar] [CrossRef] [Green Version]
- D’Esposito, V.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhou, X.; Qin, Y.; Yang, J.; Wang, Y.; Sun, Z.; Yu, K.; Zhang, S.; Liu, S. Emodin inhibits epithelial-mesenchymal transition and metastasis of triple negative breast cancer via antagonism of CC-chemokine ligand�5 secreted from adipocytes. Int. J. Mol. Med. 2018, 42, 579–588. [Google Scholar] [CrossRef]
- Fujisaki, K.; Fujimoto, H.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Shiina, N.; Kuroda, M.; Aoyagi, Y.; Miyazaki, M. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res. Treat. 2015, 150, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Danforth, D. The Role of Chronic Inflammation in the Development of Breast Cancer. Cancers 2021, 13, 3918. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1α Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Hersl, J.; La, H.; Smith, M.; Jenkins, J.; Goloubeva, O.; Dilsizian, V.; Tkaczuk, K.; Chen, W.; Jones, L. A pilot study of FDG PET/CT detects a link between brown adipose tissue and breast cancer. BMC Cancer 2014, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Pace, L.; Nicolai, E.; Basso, L.; Garbino, N.; Soricelli, A.; Salvatore, M. Brown Adipose Tissue in Breast Cancer Evaluated by [18F] FDG-PET/CT. Mol. Imaging Biol. 2020, 22, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, N.; Pepe, G.; Antunovic, L. 18F-FDG uptake of brown fat and cancer: Casualty or causality? Eur. J. Pediatr. 2019, 46, 1395–1396. [Google Scholar] [CrossRef]
- Jones, L.P.; Buelto, D.; Tago, E.; Owusu-Boaitey, K.E. Abnormal Mammary Adipose Tissue Environment of Brca1 Mutant Mice Show a Persistent Deposition of Highly Vascularized Multilocular Adipocytes. J. Cancer Sci. Ther. 2011, 01, S2–S004. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Park, S.H. Association between depression and nonalcoholic fatty liver disease: Contributions of insulin resistance and inflammation. J. Affect. Disord. 2020, 278, 259–263. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Brown, K.A. Obese Adipose Tissue as a Driver of Breast Cancer Growth and Development: Update and Emerging Evidence. Front. Oncol. 2021, 11, 638918. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Lim, W.H.; Lim, X.C.; Xiao, J.; Tan, D.J.H.; Syn, N.; Ho, C.S.H.; Kow, A.W.C.; Tan, E.X.X.; Fung, J.; et al. A meta-analysis on the incidence of donor-related depression after liver transplant. Transpl. Int. 2021, 34, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.Y.; Chang, Y.; Sung, E.; Kang, J.-H.; Wild, S.H.; Byrne, C.D.; Shin, H.; Ryu, S. Depression and increased risk of non-alcoholic fatty liver disease in individuals with obesity. Epidemiol. Psychiatr. Sci. 2021, 30, e23. [Google Scholar] [CrossRef] [PubMed]
- Orrù, M.G.; Pariante, C.M. Depression and liver diseases. Dig. Liver Dis. 2005, 37, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.; Hove, D.L.V.D.; Steinbusch, H.W.; Prickaerts, J. Major depression, cognitive dysfunction and Alzheimer’s disease: Is there a link? Eur. J. Pharmacol. 2010, 626, 72–82. [Google Scholar] [CrossRef]
- Caraci, F.; Copani, A.; Nicoletti, F.; Drago, F. Depression and Alzheimer’s disease: Neurobiological links and common pharmacological targets. Eur. J. Pharmacol. 2009, 626, 64–71. [Google Scholar] [CrossRef]
- Guo, P.; Chen, S.; Wang, H.; Wang, Y.; Wang, J. A Systematic Analysis on the Genes and Their Interaction Underlying the Comorbidity of Alzheimer’s Disease and Major Depressive Disorder. Front. Aging Neurosci. 2022, 13, 789698. [Google Scholar] [CrossRef]
- Baba, H.; Nakano, Y.; Maeshima, H.; Satomura, E.; Kita, Y.; Suzuki, T.; Arai, H. Metabolism of Amyloid-β Protein May Be Affected in Depression. J. Clin. Psychiatry 2011, 73, 115–120. [Google Scholar] [CrossRef]
- Kita, Y.; Baba, H.; Maeshima, H.; Nakano, Y.; Suzuki, T.; Arai, H. Serum amyloid β protein in young and elderly depression: A pilot study. Psychogeriatrics 2009, 9, 180–185. [Google Scholar] [CrossRef]
- Pandolfo, G.; Iannuzzo, F.; Genovese, G.; Bruno, A.; Pioggia, G.; Baldari, S.; Gangemi, S. Mental Illness and Amyloid: A Scoping Review of Scientific Evidence over the Last 10 Years (2011 to 2021). Brain Sci. 2021, 11, 1352. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Zhu, H.; Dean, M.; Liu, Z.; Vu, L.; Fan, G.; Li, H.; Mwamburi, M.; Steffens, D.C.; Au, R. Amyloid-associated depression and ApoE4 allele: Longitudinal follow-up for the development of Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2015, 31, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, D.; Zhao, J.; Song, J.; Zhao, Y. The role of the low-density lipoprotein receptor–related protein 1 (LRP-1) in regulating blood-brain barrier integrity. Rev. Neurosci. 2016, 27, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Bromek, E.; Daniel, W.A. The regulation of liver cytochrome P450 expression and activity by the brain serotonergic system in different experimental models. Expert Opin. Drug Metab. Toxicol. 2021, 17, 413–424. [Google Scholar] [CrossRef]
- Bromek, E.; Wójcikowski, J.; Daniel, W.A. Involvement of the paraventricular (PVN) and arcuate (ARC) nuclei of the hypothalamus in the central noradrenergic regulation of liver cytochrome P450. Biochem. Pharmacol. 2013, 86, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Haduch, A.; Rysz, M.; Gołembiowska, K.; Daniel, W.A. The role of brain noradrenergic system in the regulation of liver cytochrome P450 expression. Biochem. Pharmacol. 2013, 86, 800–807. [Google Scholar] [CrossRef]
- Wójcikowski, J.; Daniel, W.A. The brain dopaminergic system as an important center regulating liver cytochrome P450 in the rat. Expert Opin. Drug Metab. Toxicol. 2009, 5, 631–645. [Google Scholar] [CrossRef]
- Danek, P.; Kuban, W.; Daniel, W. The Effect of Chronic Iloperidone Treatment on Cytochrome P450 Expression and Activity in the Rat Liver: Involvement of Neuroendocrine Mechanisms. Int. J. Mol. Sci. 2021, 22, 8447. [Google Scholar] [CrossRef]
- Rifkind, A.B.; Lee, C.; Chang, T.K.; Waxman, D.J. Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: Regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch. Biochem. Biophys. 1995, 320, 380–389. [Google Scholar] [CrossRef]
- Jung, J.Y.; Park, S.K.; Oh, C.-M.; Chung, P.-W.; Ryoo, J.-H. Non-Alcoholic Fatty Liver Disease and Its Association with Depression in Korean General Population. J. Korean Med. Sci. 2019, 34, e199. [Google Scholar] [CrossRef]
- Harris, T.R.; Bettaieb, A.; Kodani, S.; Dong, H.; Myers, R.; Chiamvimonvat, N.; Haj, F.G.; Hammock, B.D. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol. Appl. Pharmacol. 2015, 286, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Cao, B.; Cheng, Q.; Cai, W.; Ye, C.; Liang, J.; Liu, W.; Tan, L.; Yan, M.; Li, B.; et al. Inhibition of soluble epoxide hydrolase ameliorates hyperhomocysteinemia-induced hepatic steatosis by enhancing β-oxidation of fatty acid in mice. Am. J. Physiol. Liver Physiol. 2019, 316, G527–G538. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.; Hsu, M.-F.; Koike, S.; Chu, B.; Cheng, J.; Yang, J.; Morisseau, C.; Torok, N.J.; Hammock, B.D.; Haj, F.G. Soluble Epoxide Hydrolase Hepatic Deficiency Ameliorates Alcohol-Associated Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 815–830. [Google Scholar] [CrossRef]
- Simopoulos, A. Importance of the Ratio of Omega-6/Omega-3 Essential Fatty Acids: Evolutionary Aspects. World Rev. Nutr. Diet. 2003, 92, 1–22. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Fabian, C.J.; Kimler, B.F.; Hursting, S.D. Omega-3 fatty acids for breast cancer prevention and survivorship. Breast Cancer Res. 2015, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflammation 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: Nutritional implications for chronic diseases. Biomed. Pharmacother. 2006, 60, 502–507. [Google Scholar] [CrossRef]
- Sellem, F.; Pesando, D.; Bodennec, G.; Girard, J.-P.; Simopoulos, A.P. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.; Stanton, C. Health Implications of High Dietary Omega-6 Polyunsaturated Fatty Acids. J. Nutr. Metab. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The Importance of the Omega-6/Omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X. The importance of omega-6/omega-3 fatty acid ratio in cell function. The gene transfer of omega-3 fatty acid desaturase. World Rev. Nutr. Diet. 2003, 92, 23–36. [Google Scholar] [PubMed]
- Simopoulos, A.P. Evolutionary Aspects of Diet: Essential Fatty Acids. World Rev. Nutr. Diet. 2001, 88, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Martinez-Urbistondo, D.; Vargas-Nuñez, J.A.; Martinez, J.A. The Role of Nutrition on Meta-inflammation: Insights and Potential Targets in Communicable and Chronic Disease Management. Curr. Obes. Rep. 2022, 22, 1–31. [Google Scholar] [CrossRef]
- Trovato, F.M.; Martines, G.F.; Catalano, D. Addressing Western dietary pattern in obesity and NAFLD. Nutrire 2018, 43, 11. [Google Scholar] [CrossRef]
- Yang, J.; Solaimani, P.; Dong, H.; Hammock, B.; Hankinson, O. Treatment of mice with 2,3,7,8-Tetrachlorodibenzo-p-dioxin markedly increases the levels of a number of cytochrome P450 metabolites of omega-3 polyunsaturated fatty acids in the liver and lung. J. Toxicol. Sci. 2013, 38, 833–836. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Kim, J.; You, M.; Giraud, D.; Toney, A.M.; Shin, S.-H.; Kim, S.-Y.; Borkowski, K.; Newman, J.W.; Chung, S. α-Linolenic acid-enriched butter attenuated high fat diet-induced insulin resistance and inflammation by promoting bioconversion of n-3 PUFA and subsequent oxylipin formation. J. Nutr. Biochem. 2019, 76, 108285. [Google Scholar] [CrossRef]
- Ansari, M.H.; Ahmad, S.; Ahmad, F.; Ahmad, M.; Osman, S.M. Co-occurrence of Coronaric and Vernolic Acids in Compositae Seed Oils. Lipid/Fett 1987, 89, 116–118. [Google Scholar] [CrossRef]
- Powell, R.G.; Smith, C.R.; Wolff, I.A. cis-5,cis-9,cis-12-octadecatrienoic and some unusual oxygenated acids inXeranthemum annuum seed oil. Lipids 1967, 2, 172–177. [Google Scholar] [CrossRef]
- Kato, T.; Yamaguchi, Y.; Uyehara, T.; Yokoyama, T.; Namai, T.; Yamanaka, S. Self defensive substances in rice plant against rice blast disease. Tetrahedron Lett. 1983, 24, 4715–4718. [Google Scholar] [CrossRef]
- Deol, S.; Fahrmann, J.; Yang, J.; Evans, J.R.; Rizo, A.; Grapov, D.; Salemi, M.; Wanichthanarak, K.; Fiehn, O.; Phinney, B.; et al. Omega-6 and omega-3 oxylipins are implicated in soybean oil-induced obesity in mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.L.; Nakamura, M.T.; Ma, D.W.L. Differentiating the biological effects of linoleic acid from arachidonic acid in health and disease. Prostaglandins Leukot. Essent. Fat. Acids 2018, 135, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bassett, J.K.; Hodge, A.M.; English, D.R.; MacInnis, R.J.; Giles, G.G. Plasma phospholipids fatty acids, dietary fatty acids, and breast cancer risk. Cancer Causes Control 2016, 27, 759–773. [Google Scholar] [CrossRef]
- Goodstine, S.L.; Zheng, T.; Holford, T.R.; Ward, B.A.; Carter, D.; Owens, P.H.; Mayne, S.T. Dietary (n-3)/(n-6) fatty acid ratio: Possible relationship to premenopausal but not postmenopausal breast cancer risk in U.S. women. J. Nutr. 2003, 133, 1409–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murff, H.J.; Shu, X.-O.; Li, H.; Yang, G.; Wu, X.; Cai, H.; Wen, W.; Gao, Y.-T.; Zheng, W. Dietary polyunsaturated fatty acids and breast cancer risk in Chinese women: A prospective cohort study. Int. J. Cancer 2010, 128, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Serna-Marquez, N.; Diaz-Aragon, R.; Reyes-Uribe, E.; Cortes-Reynosa, P.; Salazar, E.P. Linoleic acid induces migration and invasion through FFAR4- and PI3K-/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Med. Oncol. 2017, 34, 1–12. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, C.; Marcial-Medina, C.; Cervantes-Anaya, N.; Cortes-Reynosa, P.; Salazar, E.P. Migration and invasion induced by linoleic acid are mediated through fascin in MDA-MB-231 breast cancer cells. Mol. Cell. Biochem. 2017, 443, 1–10. [Google Scholar] [CrossRef]
- Villegas-Comonfort, S.; Castillo-Sanchez, R.; Serna-Marquez, N.; Cortes-Reynosa, P.; Salazar, E.P. Arachidonic acid promotes migration and invasion through a PI3K/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 169–177. [Google Scholar] [CrossRef]
- Naughton, S.S.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Linoleic acid and the pathogenesis of obesity. Prostaglandins Other Lipid Mediat. 2016, 125, 90–99. [Google Scholar] [CrossRef]
- Liu, J.; Han, L.; Zhu, L.; Yu, Y. Free fatty acids, not triglycerides, are associated with non-alcoholic liver injury progression in high fat diet induced obese rats. Lipids Health Dis. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: A cross-sectional study. Sci. Rep. 2014, 4, srep05832. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillers-Ziemer, L.E.; Kuziel, G.; Williams, A.E.; Moore, B.N.; Arendt, L.M. Breast cancer microenvironment and obesity: Challenges for therapy. Cancer Metastasis Rev. 2022, 41, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.-A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflammation 2016, 13, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Duan, J.; Yang, H.; Sun, C.; Zhong, W.; Tao, J.; Guan, X.; Jiang, H.; Hammock, B.D.; Hwang, S.H.; et al. COX-2/sEH dual inhibitor PTUPB alleviates bleomycin-induced pulmonary fibrosis in mice via inhibiting senescence. FEBS J. 2019, 287, 1666–1680. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, H.S.; Chang, S.W.; Lee, C.U.; Kim, J.S.; Jung, Y.K.; Kim, J.H.; Seo, Y.S.; Yim, H.J.; Lee, C.H.; et al. Underlying nonalcoholic fatty liver disease is a significant factor for breast cancer recurrence after curative surgery. Medicine 2019, 98, e17277. [Google Scholar] [CrossRef]
- Bilici, A.; Ozguroglu, M.; Mihmanlı, I.; Turna, H.; Adaletli, I. A case–control study of non-alcoholic fatty liver disease in breast cancer. Med. Oncol. 2007, 24, 367–371. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The Role of Cytokines in Breast Cancer Development and Progression. J. Interf. Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lengacher, C.A.; Reich, R.R.; Paterson, C.L.; Shelton, M.; Shivers, S.; Ramesar, S.; Pleasant, M.L.; Budhrani-Shani, P.; Groer, M.; Post-White, J.; et al. A Large Randomized Trial: Effects of Mindfulness-Based Stress Reduction (MBSR) for Breast Cancer (BC) Survivors on Salivary Cortisol and IL-6. Biol. Res. Nurs. 2018, 21, 39–49. [Google Scholar] [CrossRef]
- Ruffell, B.; Au, A.; Rugo, H.S.; Esserman, L.J.; Hwang, E.S.; Coussens, L.M. Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. USA 2011, 109, 2796–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Cai, D.; Ma, B.; Wu, G.; Wu, J. Skewing the Balance of Regulatory T-Cells and T-Helper 17 Cells in Breast Cancer Patients. J. Int. Med. Res. 2011, 39, 691–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, Z.M.A.; Going, J.; Edwards, J.; Elsberger, B.; Doughty, J.C.; McMillan, D. The relationship between components of tumour inflammatory cell infiltrate and clinicopathological factors and survival in patients with primary operable invasive ductal breast cancer. Br. J. Cancer 2012, 107, 864–873. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Model/Animals Used | Route of Administration | Outcome of the Study | Key Findings | References |
|---|---|---|---|---|---|
| 1 | CMS C57BL/6J mice (in vivo). | NA. | RNA-seq: 1. Hepatic Ephx2 mRNA (↑); 2. ARA metabolic pathway significantly changed. | Hepatic sEH activity was selectively altered followed by CMS. The reductions in sucrose preference and coat score that were induced by CMS were both rescued by imipramine and fluoxetine (chronic antidepressant treatment). | [64] |
| 2 | CMS C57BL/6J mice (in vivo). | NA. | Western blot: 1. Hepatic monomeric sEH (↑); 2. Hepatic oligomeric sEH (↑); 3. sEH in heart, kidney, spleen, lung, muscle, stomach, PFC, or hippocampus (-). | ||
| 3 | CMS C57BL/6J mice (in vivo). | NA. | Sucrose preference (↓); coat score (↓). | ||
| 4 | CMS C57BL/6J mice (in vivo). | Tail vein injections/AAV-Ephx2-shRNAs, AAV-NC (control)/15 days. | CMS mice injected with AAV-NC virus: 1. Anhedonia (↓), sucrose preference (↓), coat score (↓); 2. The mRNA and protein levels: hepatic sEH (↑). CMS mice injected with AAV-Ephx2-shRNAs: 1. Anhedonia (↑), sucrose preference (↑), coat score (↑); 2. The mRNA and protein levels: hepatic sEH (↓). | Hepatic sEH may play an important role in depression. | [64] |
| 5 | CMS C57BL/6J mice (in vivo). | NA. | The 1-week intervention of CMS did not affect corticosterone secretion. Corticosterone administration did not affect sEH expression in primary cultured hepatocytes. | Hepatic sEH was independent of corticosterone-related mechanisms. | [64] |
| 6 | CMS cKO mice/CMS WT mice (in vivo). | NA. | CMS-induced WT mice: sucrose preference (↓); coat score (↓); CMS-induced cKO mice: sucrose preference (↑); coat score (↑). | Hepatic deletion of Ephx2 ameliorated CMS-induced depressive phenotypes and related inflammatory response. Deletion of the hepatic Ephx2 gene reversed the CMS-induced reduction in synaptic transmission and plasticity. Deletion of the hepatic Ephx2 gene cannot affect the levels of IL-6, MCP-1, and Iba1. | [64] |
| 7 | CMS cKO mice/CMS-induced WT mice (in vivo). | NA. | CMS-induced WT mice: 1. CAMK IIα in the hippocampus (↓); CAMKIIβ in the hippocampus (↓); Arc in the hippocampus (↓); 2. Neurotrophic factors mRNA and proteins in the mPFC and the hippocampus: BDNF (↓); GDNF (↓); GFAP (↓); EAAT1 (↓); EAAT2 (↓). CMS-induced cKO mice: The hepatic deletion of Ephx2 reversed the CMS-induced downregulation. | ||
| 8 | CMS cKO mice (in vivo). | NA. | The levels in the brains of cKO mice: macrophage/microglia activity markers Iba1 mRNA (↓); cyclin-dependent kinase 11b mRNA (↓); macrophage inflammatory protein 1α mRNA (↓). | ||
| 9 | CMS C57BL/6J mice; CMS cKO mice (in vivo). | NA. | The level in the mPFC and the hippocampus: IL-6 (↑); MCP-1 (↑); Iba1 (↑). | ||
| 10 | C57BL/6J mice (in vivo). | Tail vein injections/pEf1a-Ephx2 plasmid DNA, pEf1a-eYFP (control)/detection after 3 days. | Immunohistochemistry: eYFP fluorescence was positive only in liver sections. The mRNA levels on the third day after injection: Hepatic sEH mRNA (↑); hepatic sEH oligomer levels (↑); hepatic sEH monomer levels (↑). Behavior analysis: sucrose preference (↓); immobility with forced swim test (↑). Protein expression: GluN2A in the mPFC (↓); GluN2B in the mPFC (↓). | Overexpression of hepatic Ephx2 gene-induced depressive phenotypes. | [64] |
| 11 | C57BL/6J mice (in vivo). | Tail vein injections/AAV-Ephx2, AAV-eYFP (control)/evaluation after 3 days. | The mRNA, monomer, and oligomer expression levels: hepatic sEH (↑). Behavior analysis: sucrose preference (↓); immobility with forced swim test (↑). | ||
| 12 | cKO mice (in vivo). | NA. | Female cKO mice: plasma levels of 14, 15-EET (↑). Male cKO mice: plasma levels of 14, 15-EET (↑). | 14, 15-EET production is specifically regulated by the liver. 14, 15-EET plasma levels are positively correlated with sucrose preference and coat scores. 14, 15-EET plasma levels are positively correlated with sucrose preference. | [64] |
| 13 | CMS mice (in vivo). | NA. | Plasma levels of 14, 15-EET (↓). | ||
| 14 | CMS mice; cKO mice (in vivo). | Tail vein injection/AAV-Ephx2, pEf1a-Ephx2/evaluation after 4 weeks. | Female mice and the mice injected with AAV-Ephx2 or pEf1a-Ephx2: plasma levels of 14, 15-EET (↓). cKO mice: plasma levels of 14, 15-EET (-). | ||
| 15 | cKO mice; WT littermates (in vivo). | i.c.v. infusions of 14,15-EEZE/3 weeks; a cannula was implanted in the right ventricle of cKO mice or WT littermates. | cKO mice: The antidepressant-like effect (↓) after 30 min infusions. Behavior analysis: sucrose preference (-); immobility with forced swim test (-). | ||
| 16 | Neurons were isolated from the mPFC or the hippocampus of neonatal C57BL/6J mice (in vivo). | 14, 15-EET/3 h. | The mRNA levels of synaptic proteins and neurotrophic factors (-). | ||
| 17 | Neurons; astrocytes (in vitro). | Coculture. | Hippocampal neurons mRNA levels: Grin2a mRNA (↑), Grin2b mRNA (↑), Gria1 (↑), Gria2 (↑), Camk2a (↑), Camk2b (↑), Arc (↑), Bdnf (↑), Gdnf (↑), Ngf (↑), Vegfa (↑), Vegfb (↑). | Astrocytic EET signaling in the mPFC mediated the effects of hepatic Ephx2 deletion. sEH is expressed primarily in astrocytes in the brain, and astrocyte EET can bind and regulate blood flow to neuronal activity. Astrocytes are the target cells for exogenous 14, 15-EET. | [64] |
| 18 | C57BL/6J mice (in vivo). | Infusing into mPFC or the hippocampus via a cannula; 14, 15-EET; evaluation after 30 min. | Infusion into mPFC: immobility (↓), locomotor activity (-). Infusion into the hippocampus: immobility (-), locomotor activity (-). | ||
| 19 | cKO mice; WT littermates (in vivo). | Lenti-hEPHX2 injection Lenti-EGFP (control). | Lenti-hEPHX2 injection in the mPFC: sEH (↑); CMS cKO mice injected with Lenti-EGFP: resilience (↑); Lenti-hEPHX2 injection in the mPFC of WT littermates: anhedonia (↑), sucrose preference (↓), and coat deterioration (↑). The antidepressant-like effect induced by the hepatic deletion of Ephx2 was blocked by the overexpression of hEPHX2 in the mPFC. The increased protein levels of GluA1, GluA2, and GluN2A in the mPFC of cKO mice were attenuated by the overexpression of hEPHX2 in the mPFC. | ||
| 20 | Male C57BL/6 mice (8–10 weeks old); model: FST (in vivo). | 0.01, 0.1, 1 mg/kg TPPU (sEHI)/i.p./30 min. | 0.01, 0.1 mg/kg TPPU and 1 mL imipramine reduce immobility time in mice during FST. | Acute administration of sEHI TPPU decreases depressant phenotypes. Intraperitoneal administration of TPPU for seven days also reverses the depressant phenotypes. | [59] |
| 21 | Male C57BL/6 mice (8–10 weeks old); model: NSF (in vivo). | 0.01, 0.1, 1 mg/kg TPPU (sEHI)/i.p./30 min. | 1. Feed latency (↓); 2. Improvement in antidepressant phenotypes and cell proliferation were inhibited by BDNF–trkB antagonist K252a; 3. Hippocampal BDNF expression (↑), cell proliferation in the dentate gyrus (↑). | ||
| 22 | Female and male mice in which microglia expressed the green fluorescent protein (GFP; fms-EGFP or MacGreen mice) (in vivo). | A single intraperitoneal injection of LPS (1 mg/kg). | 1. Neuroblast number (↓), whose effect was exacerbated by the ω-3 PUFA-deficient diet; 2. The ω-3 PUFA-deficient diet reduced the DG volume, AHN, microglia number, and surveilled volume; 3. The diet effect on most mature neuroblasts was exclusively significant in female mice. | Colocalization and multivariate analysis revealed an association between microglia and AHN, as well as the sexual dimorphic effect of diet. Female mice are more susceptible than males to the effect of dietary ω-6/ω-3 PUFA ratio on AHN and microglia. | [65] |
| 23 | MD Wistar rats; RS Wistar rats; MDR Wistar rats (in vivo). | NA. | Plasma corticosterone levels: RS rats (↑), MD rats (↑); CYP3A2 expression at the mRNA, apoprotein, and activity (6β-testosterone hydroxylation) level: RS rats (↑), MD rats (↑), MDR rats (↑); CYP3A1 expression at the mRNA, apoprotein, and activity (6β-testosterone hydroxylation) level: MD rats (↑), MDR rats (↑); CYP2C11 expression at the mRNA and activity level: MDR rats (↑); CYP2D1 expression at the mRNA, apoprotein, and activity (6β-testosterone hydroxylation) level: RS rats (↑); CYP2D1 expression at the mRNA: MDR rats (↑). | 1. MD and RS regulate CYP expression. 2. The expression of CYP3A1 and CYP2C11 was increased in the liver of MD rats, whereas RS had no significant effect. 3. Hepatic CYP2D1/2 activity was increased by RS, whereas MD did not affect it. | [66] |
| 24 | Wistar rats (in vivo). | Phenylephrine hydrochloride, 2 mg/kg i.p., 1 × 4, α1-agonist. | CYP3A1 transcripts (↑), CYP3A2 expression (↑). | ||
| 25 | Primary hepatocytes (in vitro). | Corticosterone (CORT; 1–25 μM). | CYP2C11 transcripts (↑), CYP2D2 transcripts (↑). | ||
| 26 | Primary hepatocytes (in vitro). | Epinephrine (10 μM, 24 h). | CYP3A1 expression (↑), CYP3A2 expression (↑), CYP2C11 transcripts (↑). |
| S. No | Samples | Outcome of the Study | Key Findings | References |
|---|---|---|---|---|
| 1 | Blood plasma from MDD patients. | ELISA: 14, 15-EET (↑); 5, 6-EET (-); 8, 9-EET (-); 11, 12-EET (-). | 1. A change in the ratio of epoxides to the corresponding diols in the plasma, including EET/DHET, may be an indicator of the activity of hepatic sEH. 2. The conversion of EET to DHET by sEH is regioselective, and 14, 15-EET is the preferred substrate. 3. The enzymatic activity of sEH in the liver was altered in patients with MDD. | [64] |
| 2 | Blood plasma from MDD patients. | ELISA: 14, 15-DHETs (↑); 5, 6-DHET (-); 8, 9-DHET (-); 11, 12-DHET (-). | ||
| 3 | Blood plasma from MDD patients. | ELISA: Total ratio of EETs/DHETs (↓); Ratio of 14, 15-EET/14, 15-DHET (↓). | ||
| 4 | Human postmortem parietal cortex (Brodmann area 7) from MDD patients, BD patients, and SZ patients. | Western blot: sEH in the parietal cortex from MDD, BD, and SZ groups (↑). | 1. There was a positive correlation between sEH protein in the parietal cortex and hepatic sEH. 2. The increased expression of sEH in the brain and hepatic sEH might play a role in the pathogenesis of major psychiatric disorders, 3. Brain-liver axis may be a critical part in major psychiatric disorders. | [67] |
| 5 | Human liver from MDD patients, BD patients, and SZ patients. | Western blot: sEH in the liver from MDD, BD, and SZ groups (↑). |
| S. No | Model/Animals/Cells Used | Administration | Outcome of the Study | Key Findings | References |
|---|---|---|---|---|---|
| 1 | MCF-7 cells (in vitro). | 9,10- and 12,13-DiHOME (0.32–1.6 μM). | Cell proliferation (↑). | Mechanisms not involving estrogen receptor or nuclear type II binding sites. | [26] |
| 2 | MDA-MB-231 breast cancer cells (in vitro). | Stimulation with AA was performed with a solution of AA dissolved in ethanol. | p-Akt (↑); Migration (↑); Invasion (↑). | Akt/PI3K and EGFR pathways mediate migration and invasion induced by AA in MDA-MB-231 breast cancer cells. | [149] |
| 3 | MDA-MB-231 cells (in vitro). | DMEM/F12 (1:1) supplemented with 5% FBS, 10 lg/mL insulin, 0.5 lg/mL hydrocortisone, 20 ng/mL EGF, and antibiotics. | p-Akt (↑); FFAR (↑); NF-κB (↑). | LA induces migration and invasion through an EGFR/PI3K/Akt-dependent pathway in breast cancer cells. | [147] |
| 4 | Male C57BL/6 N mice (in vivo). | 1. CO; 2. The conventional soybean oil diet contains 50% CO and 50% SO (SO + CO); 3. The PL + CO diet. | SO+CO group mice: hepatic AA (↑), plasma AA (↑); hepatic LA (↑), plasma LA (↑); hepatic 9, 10-DiHOME (↑), hepatic 12, 13-DiHOME (↑). | Increased LA consumption leads to increased concentrations of EpOMEs and DiHOMEs in the liver, which cause the upregulation of 9, 10-DiHOME. | [142] |
| 5 | Male C57BL/6J mice (in vivo). | A commercially available butter blend (Hiland Dairy Foods); trans-fat free margarine (Land O’Lakes); ALA-enriched butter (Sunseo Milk Butter™). | Dietary reduction in ω-6/ω-3 FA ratio is effective in reducing systemic levels of ω-6/ω-3 ratio, promoting biosynthesis of long-chain ω-3 PUFA and generation of both ALA- and EPA-derived oxylipins. | Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. | [154] |
| 6 | Sf-21 cells infected with recombinant baculoviruses produce either hsEH, msEH, hmEH, or lacZ (in vitro). | 2% final concentration of DMSO (vol/vol)/3 days. | The toxicity of the leukotoxins in cells expressing msEH could be reversed by the administration of the potent sEH inhibitor 4-fluorochalcone oxide. | The numerous pathologies attributed to leukotoxin and isoleukotoxin result from enzymatic activation mediated largely by the soluble epoxide hydrolase. | [73] |
| 7 | Sprague Dawley rats (in vivo). | Free fatty acids dissolved in PBS containing 5% DMSO by cardiac puncture after anesthetized by intraperitoneal injections of pentobarbital. | 35 mg/kg leukotoxin diol caused immediate respiratory distress with 100% death in less than 2 h. | The leukotoxins can be metabolism by EHs in target tissues or the diols can be formed in hepatic and renal tissues with high sEH and then released into general circulation. | |
| 8 | MDA-MB-231 breast cancer cells (in vitro). | LA. | 1. The formation of filopodia and lamellipodia (↑); 2. The localization of fascin (↑); 3. Migration (↑), invasion (↑), matrix metalloproteinase-9 secretion (↑). | LA induces the formation of filopodia and lamellipodia and the localization of fascin in these actin structures in MDA-MB-231 breast cancer cells. Fascin is required for migration and invasion induced by LA in MDA-MB-231 breast cancer cells. | [149] |
| 9 | MCF12A breast cancer cells (in vitro). | LA. | 1. The formation of microspikes (↑); 2. The localization of fascin (↑). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Ganesan, K.; Wu, M.; Hu, Y.; She, Y.; Tian, Q.; Ye, Q.; Chen, J. Crosstalk between Depression and Breast Cancer via Hepatic Epoxide Metabolism: A Central Comorbidity Mechanism. Molecules 2022, 27, 7269. https://doi.org/10.3390/molecules27217269
Ye Z, Ganesan K, Wu M, Hu Y, She Y, Tian Q, Ye Q, Chen J. Crosstalk between Depression and Breast Cancer via Hepatic Epoxide Metabolism: A Central Comorbidity Mechanism. Molecules. 2022; 27(21):7269. https://doi.org/10.3390/molecules27217269
Chicago/Turabian StyleYe, Zhen, Kumar Ganesan, Mingquan Wu, Yu Hu, Yingqi She, Qianqian Tian, Qiaobo Ye, and Jianping Chen. 2022. "Crosstalk between Depression and Breast Cancer via Hepatic Epoxide Metabolism: A Central Comorbidity Mechanism" Molecules 27, no. 21: 7269. https://doi.org/10.3390/molecules27217269