
Synthesis of Cyclotetrapeptides Analogues to Natural Products as Herbicides

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
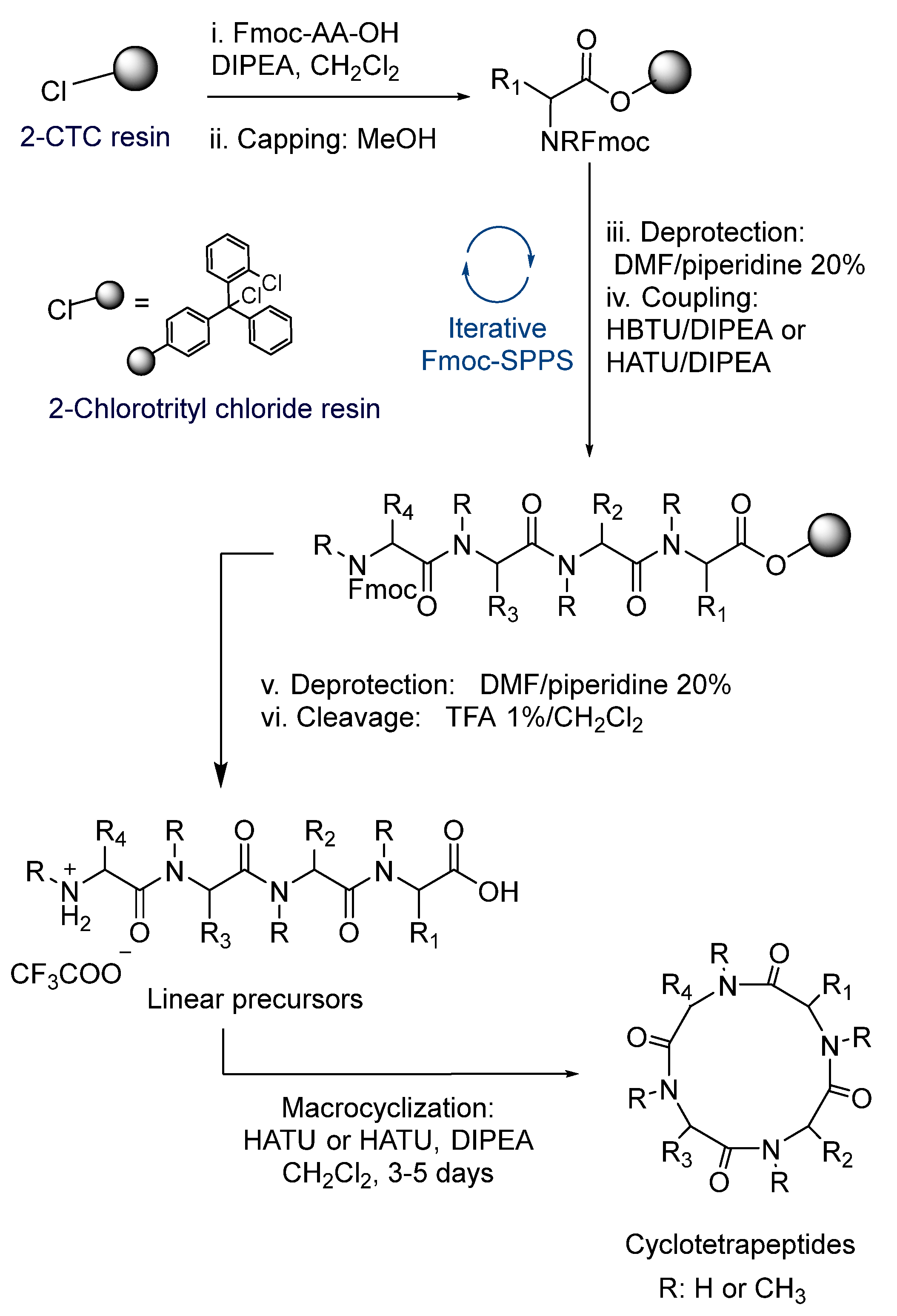
2.1. Synthesis
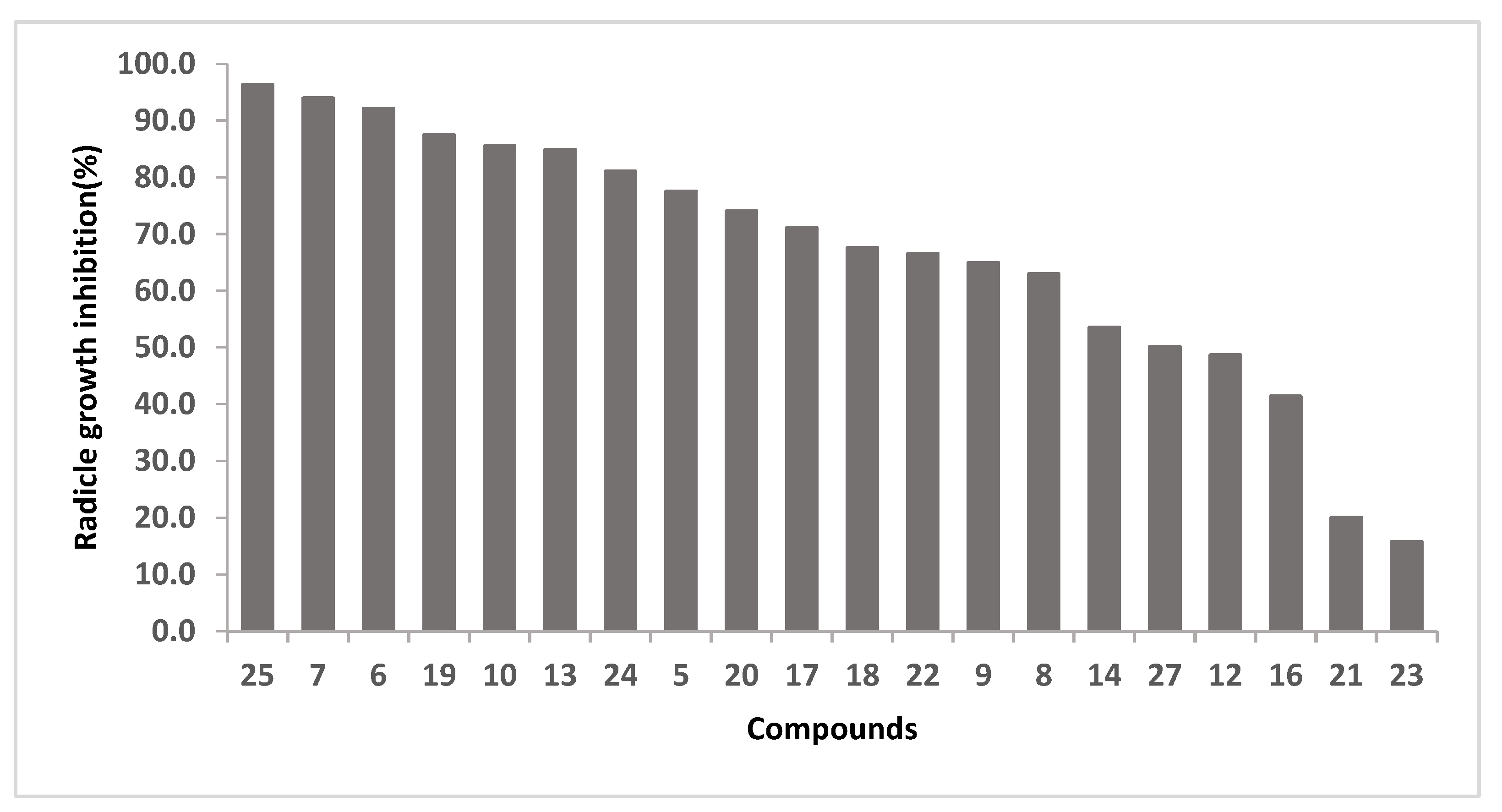
2.2. Herbicidal Activity
3. Materials and Methods
3.1. Synthesis
- (I)
- Resin loading: 500 mg of 2-Chlorotrityl chloride resin (2-CTC) were added to a plastic syringe. The resin was swelled in CH2Cl2 (3 × 5 min). A solution of first protected amino acid Fmoc-AA-OH (1 eq. for 0.8 mmol/g loading) and DIPEA (3 eq.) in CH2Cl2 was added and shaken for 10 min. Then, an extra 7.0 eq. of DIPEA were added, and shaking was continued for 50 min. MeOH (0.8 mL/g of resin) was added to the previous mixture and shaken for 10 min. After filtering, the resin was washed with CH2Cl2 (×3), MeOH (×3), CH2Cl2 (×3), DMF (×3).
- (II)
- Removal of NHFmoc group: The resin was shaken with piperidine-DMF solution (1:4) (1 × 1 min and 2 × 5 min). In exceptional cases, deprotection step was accomplished by a single treatment with piperidine-DMF solution for 5 min in order to prevent side reactions.
- (III)
- Coupling of subsequent N-Fmoc protected amino acids to primary or secondary amines: After removal of NHFmoc-protecting group, as previously described, the resin was washed with DMF (×3), CH2Cl2 (×3) and DMF (×3). Then, a solution of Fmoc-AA-OH (3 eq.) and DIPEA (6 eq.) in DMF was added to the resin, followed by a solution of HBTU, for coupling to primary amines, or HATU (2.9 eq.) in DMF, in case of coupling to an N-methylamino acid. The mixture was stirred for 60 min. After the coupling was completed, the resin was washed with DMF (×3) and CH2Cl2 (×3). Successive deprotection and coupling cycles were made with the appropriate amino acids to obtain the desired compound. The coupling was monitored by colorimetric assays; Kaiser test in case of primary amines and Chloranil test for secondary amines. Coupling procedure was repeated in case of positive results.
- (IV)
- Cleavage: The peptide was cleaved from the resin by treatment with 1% TFA in CH2Cl2 for 2–3 min at room temperature, followed by filtration and collection of the filtrate in MeOH. The treatment was repeated three times and then the resin was washed with CH2Cl2 (×5) and MeOH (×3). Solvents were removed under vacuum to obtain the crude peptide. LC-MS was used to identify the desired product
- (V)
- General procedure for macrocyclization in solution phase to obtain (16–27):Method I: Macrocyclization reaction was performed in diluted conditions (1–5 mM) using HBTU or HATU (1.5 eq.), DIPEA (3 eq.), 4-DMAP (catalytic) in dried CH2Cl2 at room temperature during 1–5 days. The reaction mixture was washed with HCl 5%, later with saturated aqueous NaHCO3, dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography to obtain the pure macrocycle.Method II: The trifluoroacetate salt of the corresponding linear peptide was dissolved in dried CH2Cl2 and diluted to a concentration of 1–5 mM. DIPEA (1 eq.) was added to enable dissolution. EDCI (1.2 eq) and oxyma (1.2 eq.) were added at 0 °C and the reaction mixture was stirred for 10 min. Then, the reaction mixture is allowed to reach room temperature and stirred for 48 h. The reaction mixture was washed with HCl 5%, later with saturated aqueous NaHCO3, dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography to obtain the pure macrocycle.
3.2. Herbicidal Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, A.; Kumar, V.; Shahzad, B.; Tanveer, M.; Sidhu, G.P.S.; Handa, N.; Kohli, S.K.; Yadav, P.; Bali, A.S.; Parihar, R.D.; et al. Worldwide Pesticide Usage and Its Impacts on Ecosystem. SN Appl. Sci. 2019, 1, 1446. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.M.; Sandrini, J.Z.; Souza, M.M. Toxicity Induced by Glyphosate and Glyphosate-Based Herbicides in the Zebrafish Hepatocyte Cell Line (ZF-L). Ecotoxicol. Environ. Saf. 2018, 162, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Peillex, C.; Pelletier, M. The Impact and Toxicity of Glyphosate and Glyphosate-Based Herbicides on Health and Immunity. J. Immunotoxicol. 2020, 17, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Meftaul, I.M.; Venkateswarlu, K.; Dharmarajan, R.; Annamalai, P.; Asaduzzaman, M.; Parven, A.; Megharaj, M. Controversies over Human Health and Ecological Impacts of Glyphosate: Is It to Be Banned in Modern Agriculture? Environ. Pollut. 2020, 263, 114372. [Google Scholar] [CrossRef]
- Wheeler, W.B. Role of Research and Regulation in 50 Years of Pest Management in Agriculture Prepared for the 50th Anniversary of the Journal of Agricultural and Food Chemistry. J. Agric. Food Chem. 2002, 50, 4151–4155. [Google Scholar] [CrossRef]
- Huisman, J.; Codd, G.A.; Paer, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef]
- Carmichael, W.W. Health Effects of Toxin-Producing Cyanobacteria: “The CyanoHABs”. Hum. Ecol. Risk Assess. Int. J. 2001, 7, 1393–1407. [Google Scholar] [CrossRef]
- Heap, I. Global Perspective of Herbicide-Resistant Weeds. Pest Manag. Sci. 2014, 70, 1306–1315. [Google Scholar] [CrossRef]
- Peterson, M.A.; Collavo, A.; Ovejero, R.; Shivrain, V.; Walsh, M.J. The Challenge of Herbicide Resistance around the World: A Current Summary. Pest Manag. Sci. 2018, 74, 2246–2259. [Google Scholar] [CrossRef]
- Green, J.M.; Owen, M.D.K. Herbicide-Resistant Crops: Utilities and Limitations for Herbicide-Resistant Weed Management. J. Agric. Food Chem. 2011, 59, 5819–5829. [Google Scholar] [CrossRef]
- Davis, A.S.; Frisvold, G.B. Are Herbicides a Once in a Century Method of Weed Control? Pest Manag. Sci. 2017, 73, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O. Why Have No New Herbicide Modes of Action Appeared in Recent Years? Pest Manag. Sci. 2012, 68, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B.; Strek, H.J. Herbicide Discovery in Light of Rapidly Spreading Resistance and Ever-Increasing Regulatory Hurdles. Pest Manag. Sci. 2018, 74, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Seiber, J.N.; Coats, J.; Duke, S.O.; Gross, A.D. Biopesticides: State of the Art and Future Opportunities. J. Agric. Food Chem. 2014, 62, 11613–11619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vurro, M.; Boari, A. Natural Compounds for Novel Strategies of Parasitic Plant Management. In Natural Products for Pest Management; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2006; Volume 927, pp. 76–87. [Google Scholar] [CrossRef]
- Evidente, A. Chemical and Biological Characterization of Toxins Produced by Weed Pathogenic Fungi as Potential Natural Herbicides. In Natural Products for Pest Management; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2006; Volume 927, pp. 62–75. [Google Scholar] [CrossRef]
- Hasan, M.; Ahmad-Hamdani, M.S.; Rosli, A.M.; Hamdan, H. Bioherbicides: An Eco-Friendly Tool for Sustainable Weed Management. Plants 2021, 10, 1212. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
- Staderini, M.; Megia-Fernandez, A.; Dhaliwal, K.; Bradley, M. Peptides for Optical Medical Imaging and Steps towards Therapy. Bioorg. Med. Chem. 2018, 26, 2816–2826. [Google Scholar] [CrossRef]
- Brea, R.J.; Reiriz, C.; Granja, J.R. Towards Functional Bionanomaterials Based on Self-Assembling Cyclic Peptide Nanotubes. Chem. Soc. Rev. 2010, 39, 1448–1456. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef]
- Sarojini, V.; Cameron, A.J.; Varnava, K.G.; Denny, W.A.; Sanjayan, G. Cyclic Tetrapeptides from Nature and Design: A Review of Synthetic Methodologies, Structure, and Function. Chem. Rev. 2019, 119, 10318–10359. [Google Scholar] [CrossRef]
- Martì-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lax, A.R.; Shepherd, H.S.; Edwards, J.V. Tentoxin, A Chlorosis-Inducing Toxin from Alternaria as a Potential Herbicide. Weed Technol. 1988, 2, 540–544. [Google Scholar] [CrossRef]
- Hessel-Pras, S.; Kieshauer, J.; Roenn, G.; Luckert, C.; Braeuning, A.; Lampen, A. In Vitro Characterization of Hepatic Toxicity of Alternaria Toxins. Mycotoxin Res. 2019, 35, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Rich, D.H.; Mathiaparanam, P. Synthesis of the Cyclic Tetrapeptide Tentoxin. Effect of an N-Methyldehydrophenylalanyl Residue on Conformation of Linear Tetrapeptides. Tetrahedron Lett 1974, 15, 4037–4040. [Google Scholar] [CrossRef]
- Jiménez, J.C.; Chavarría, B.; López-Macià, À.; Royo, M.; Giralt, E.; Albericio, F. Tentoxin as a Scaffold for Drug Discovery. Total Solid-Phase Synthesis of Tentoxin and a Library of Analogues. Org. Lett. 2003, 5, 2115–2118. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, N.; Cavelier, F.; Noel, J.-P.; Gomis, J.-M. High Yield Synthesis of Tentoxin, a Cyclic Tetrapeptide. J. Pept. Sci. 2002, 8, 335–346. [Google Scholar] [CrossRef]
- Cavelier, F.; Verducci, J. New Synthesis of the Cyclic Tetrapeptide Tentoxin Employing an Azlactone as Key Intermediate. Tetrahedron Lett. 1995, 36, 4425–4428. [Google Scholar] [CrossRef]
- Edwards, J.V.; Lax, A.R.; Lillehoj, E.B.; Boudreaux, G.J. New Synthesis and Biological Activity of the Cyclic Tetrapeptides Tentoxin and [Pro 1] Tentoxin. Int. J. Pept. Protein Res 1986, 28, 603–612. [Google Scholar] [CrossRef]
- Posada, L.; Serra, G. First Total Synthesis of Versicotide D and Analogs. Tetrahedron Lett. 2019, 60, 151281. [Google Scholar] [CrossRef]
- Posada, L.; Rey, L.; Villalba, J.; Colombo, S.; Aubriot, L.; Badagian, N.; Brena, B.; Serra, G. Cyclopeptides Natural Products as Herbicides and Inhibitors of Cyanobacteria: Synthesis of Versicotides E and F. Chem. Sel. 2022, 7, e202201956. [Google Scholar] [CrossRef]
- Barlos, K.; Gatos, D.; Schäfer, W. Synthesis of prothymosin a (ProTα)—A protein consisting of 109 amino acid residues. Angew. Chem. Int. Ed. Engl. 1991, 30, 590. [Google Scholar] [CrossRef]
- Schmidt, U.; Langner, J. Cyclotetrapeptides and cyclopentapeptides: Occurrence and synthesis. J. Pep. Res. 1997, 49, 67. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yudin, A. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1988. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Compound | Yield (%) | Purity (%) * |
|---|---|---|---|
| Ala-Leu-d-Phe-Gly (4) | 4 | 100 | ND |
| N-MeAla-Leu-Phe-Gly (5) | 5 | 100 | 93 |
| Ala-Leu-Phe-N-MeGly (6) | 6 | 60 | 94 |
| Ala-Leu-N-Me-d-Phe-Gly (7) | 7 | 100 | 99 |
| N-MeAla-Leu-d-Phe-Gly (8) | 8 | 100 | 96 |
| Ala-Leu-d-Phe-N-MeGly (9) | 9 | 62 | 90 |
| Ala-Leu-N-MePhe-N-MeGly (10) | 10 | 100 | 97 |
| N-MeAla-Leu-Phe-N-MeGly (11) | 11 | 44 | 94 |
| N-MeAla-Leu-N-MePhe-Gly (12) | 12 | 100 | 91 |
| N-MeAla-Phe-N-MePhe-Gly (13) | 13 | 97 | 98 |
| N-Me-d-Phe-Ala-Phe-N-MeGly (14) | 14 | 99 | 99 |
| Phe-N-MeGly-Cys(Bn)-N-MeGly (15) | 15 | 75 | 79 |
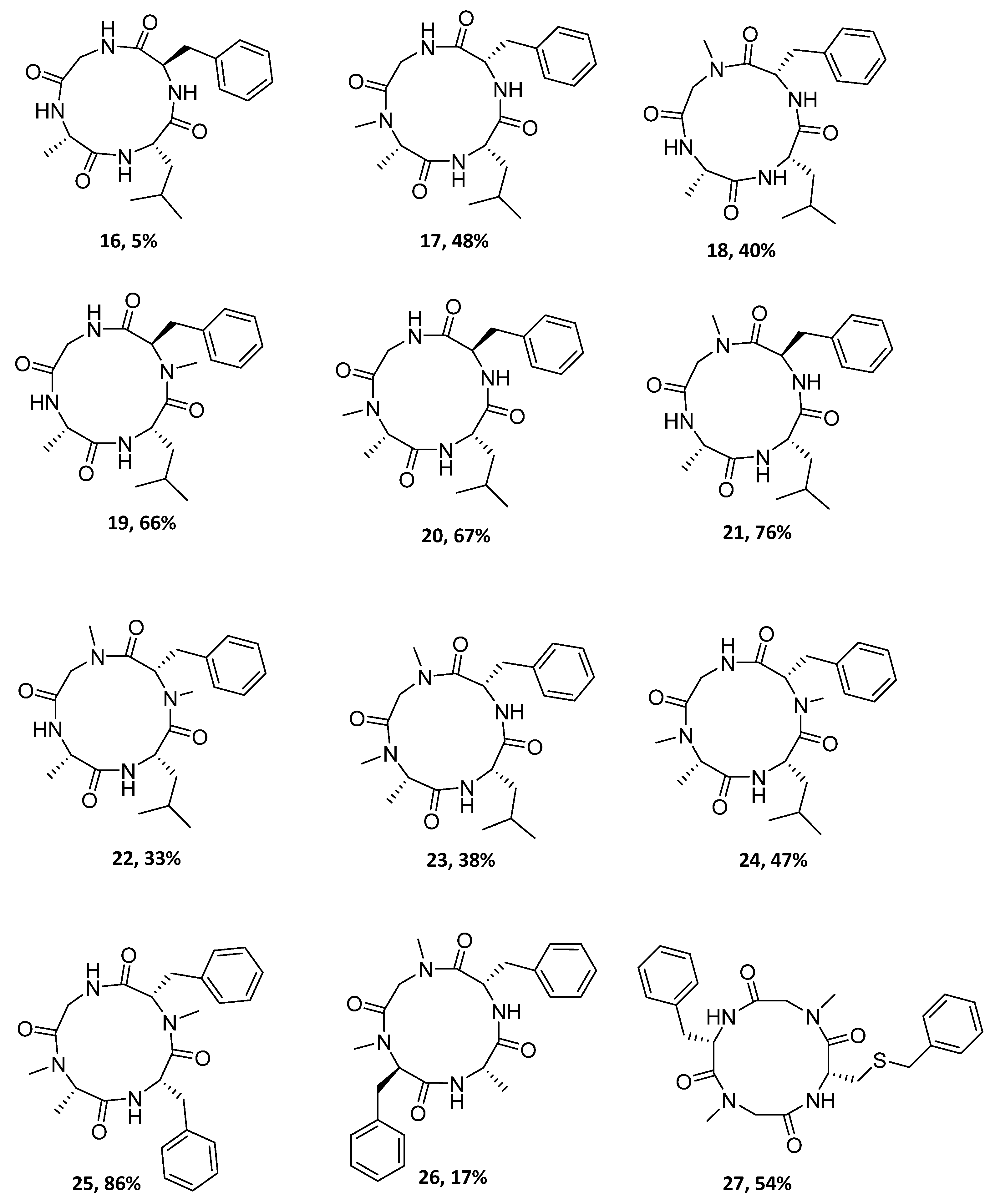
| Peptide Precursor | Cyclopeptide | Macrocyclization Yield (%) |
|---|---|---|
| Ala-Leu-d-Phe-Gly (4) | 16 | 5 |
| N-MeAla-Leu-Phe-Gly (5) | 17 | 48 |
| Ala-Leu-Phe-N-MeGly (6) | 18 | 40 |
| Ala-Leu-NMe-d-Phe-Gly (7) | 19 | 66 |
| N-MeAla-Leu-d-Phe-Gly (8) | 20 | 67 |
| Ala-Leu-d-Phe-N-MeGly (9) | 21 | 76 |
| Ala-Leu-N-MePhe-N-MeGly (10) | 22 | 33 |
| N-MeAla-Leu-Phe-N-MeGly (11) | 23 | 38 |
| N-MeAla-Leu-N-MePhe-Gly (12) | 24 | 47 |
| N-MeAla-Phe-N-MePhe-Gly (13) | 25 | 86 |
| N-Me-d-Phe-Ala-Phe-N-MeGly (14) | 26 | 17 |
| Phe-N-MeGly-Cys(Bn)-N-MeGly (15) | 27 | 54 |
| Compound | Concentration (µM) | Radicle Growth Inhibition (%) |
|---|---|---|
| 6 | 23 | No inhibition |
| 7 | 11 | 48 |
| 10 | 11 | 15 |
| 13 | 5 | 38 |
| 17 | 6 | 10 |
| 19 | 9 | 53 |
| 20 | 6 | 32 |
| 24 | 10 | 62 |
| 25 | 4 | 41 |
| 26 | 7 | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irabuena, C.; Posada, L.; Rey, L.; Scarone, L.; Davyt, D.; Villalba, J.; Serra, G. Synthesis of Cyclotetrapeptides Analogues to Natural Products as Herbicides. Molecules 2022, 27, 7350. https://doi.org/10.3390/molecules27217350
Irabuena C, Posada L, Rey L, Scarone L, Davyt D, Villalba J, Serra G. Synthesis of Cyclotetrapeptides Analogues to Natural Products as Herbicides. Molecules. 2022; 27(21):7350. https://doi.org/10.3390/molecules27217350
Chicago/Turabian StyleIrabuena, Camila, Laura Posada, Luciana Rey, Laura Scarone, Danilo Davyt, Juana Villalba, and Gloria Serra. 2022. "Synthesis of Cyclotetrapeptides Analogues to Natural Products as Herbicides" Molecules 27, no. 21: 7350. https://doi.org/10.3390/molecules27217350