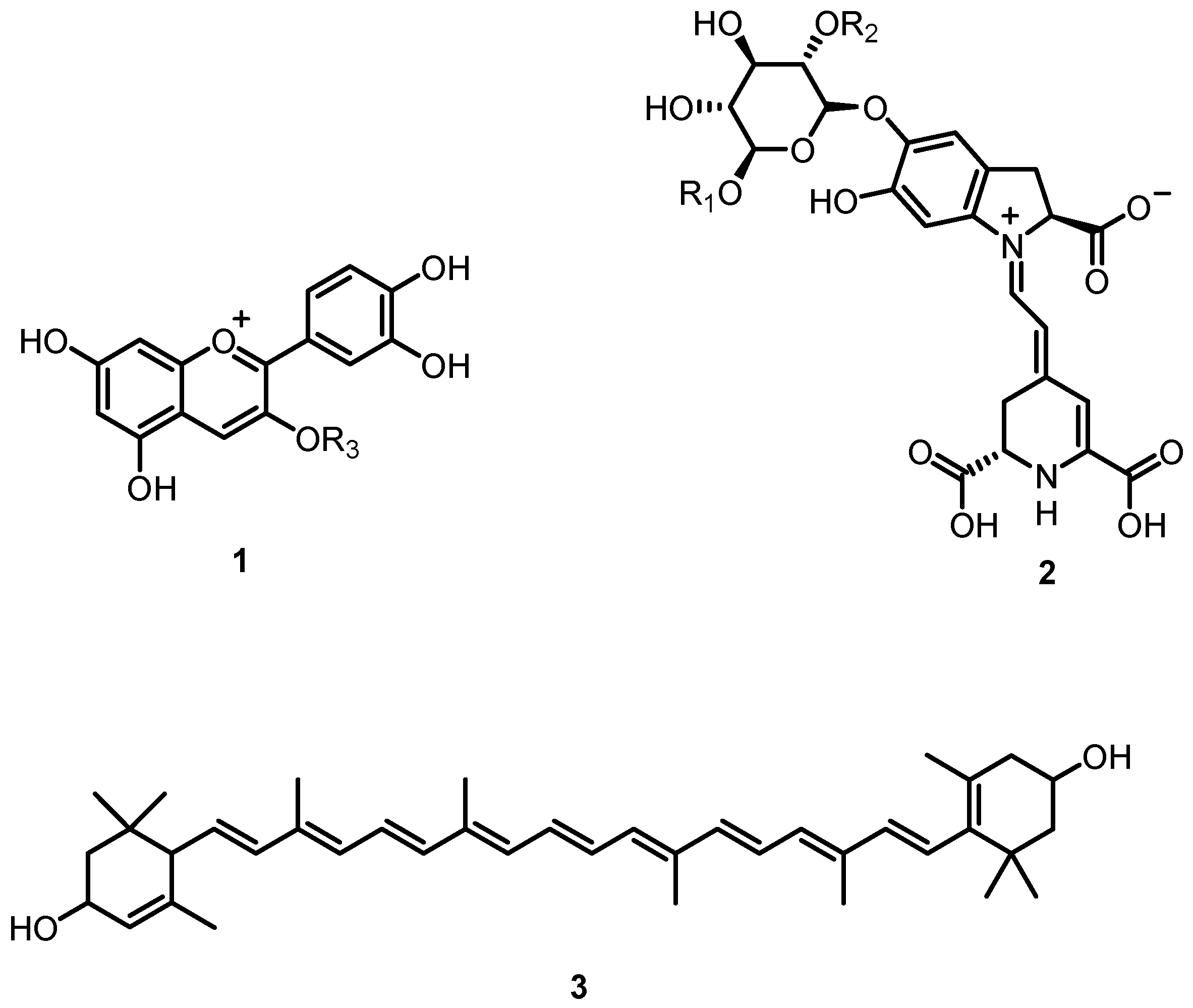
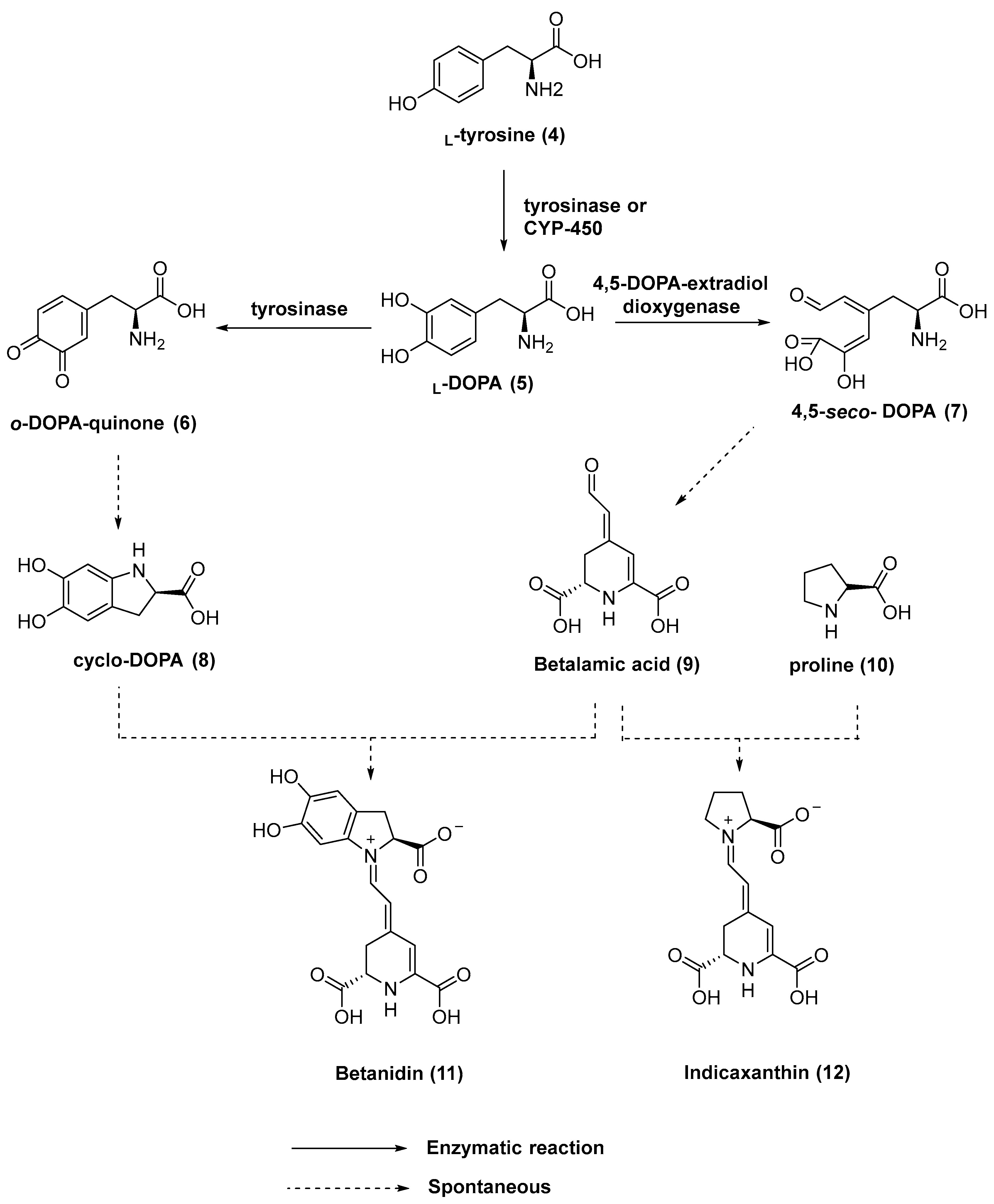
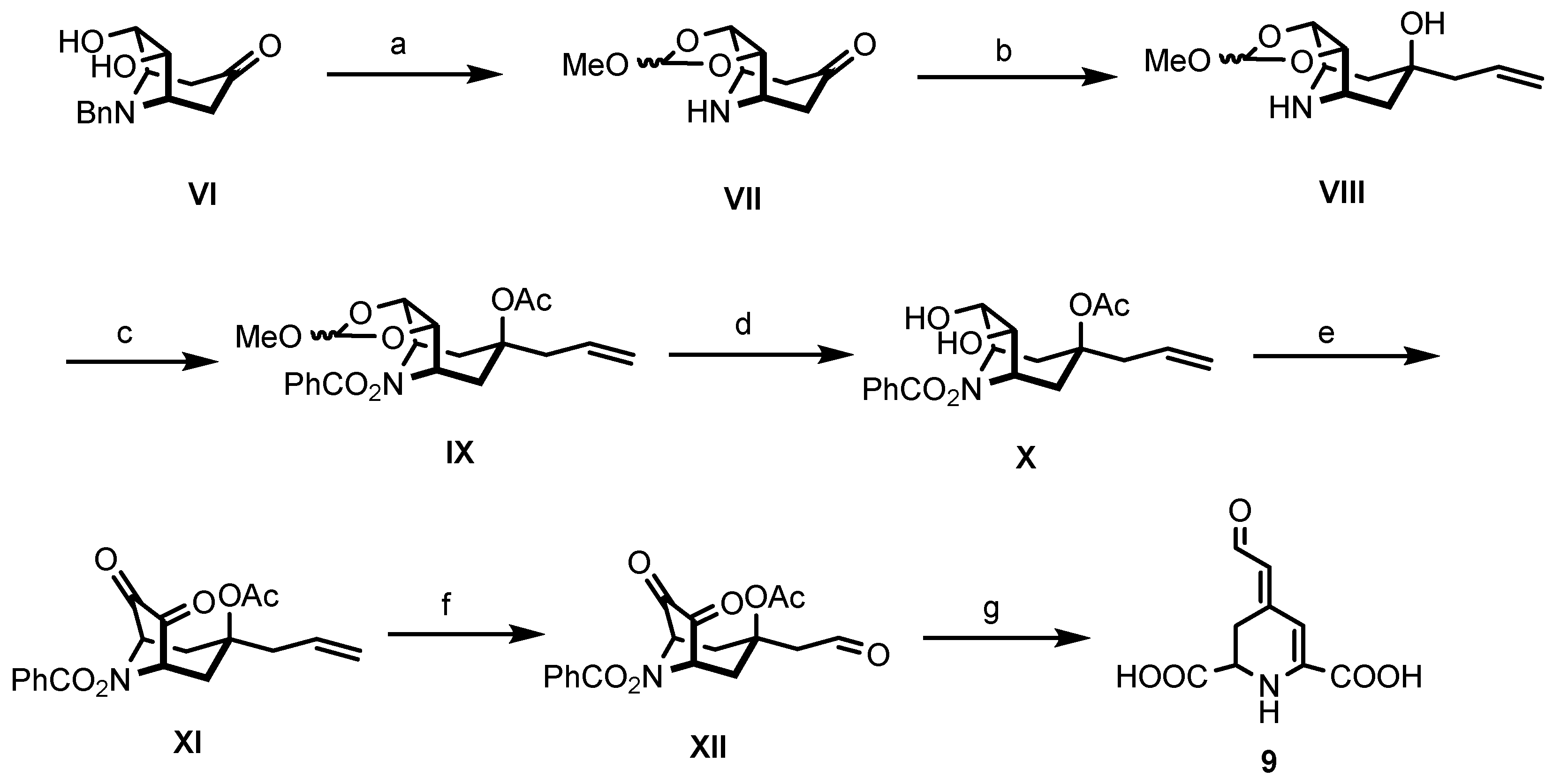
3.1. Chemistry
Moisture and oxygen sensitive reactions were carried out under nitrogen, dried with molecular sieves (3 or 4 Å, 8 to 12 mesh, Acros Organics), in dry glassware (Schlenk flasks or Schlenk tubes, sealed with rubber septa). All solvents were of analytical grade quality. Flash chromatography (FC): silica gel 60, 40–63 µm (Machery Nagel); parentheses include: diameter of the column (
Ø), length of the stationary phase (h), fraction size (
V) and eluent. Melting point: melting point system MP50 (Mettler Toledo, gießen, Germany), open capillary, uncorrected. MS: MicroTOFQII mass spectrometer (Bruker Daltonics, Bremen, Germany); deviations of the found exact masses from the calculated exact masses were 5 ppm or less; the data were analyzed with DataAnalysis
® (Bruker Daltonics). NMR: NMR spectra were recorded in deuterated solvents on Agilent DD2 400 MHz and 600 MHz NMR spectrometers (Agilent, Santa Clara, CA, USA); chemical shifts (
δ) are reported in parts per million (ppm) against the reference substance tetramethylsilane and calculated using the solvent residual peak of the undeuterated solvent; coupling constants are given with 0.5 Hz resolution; assignment of
1H and
13C NMR signals was supported by 2D NMR techniques where necessary. IR: FT/IR IR Affinity
®-1 spectrometer (Shimadzu, Düsseldorf, Germany) using ATR technique. Characterization data including
1H and
13C NMR spectra for synthesized compounds are reported in
Supplementary Materials (Page S3).
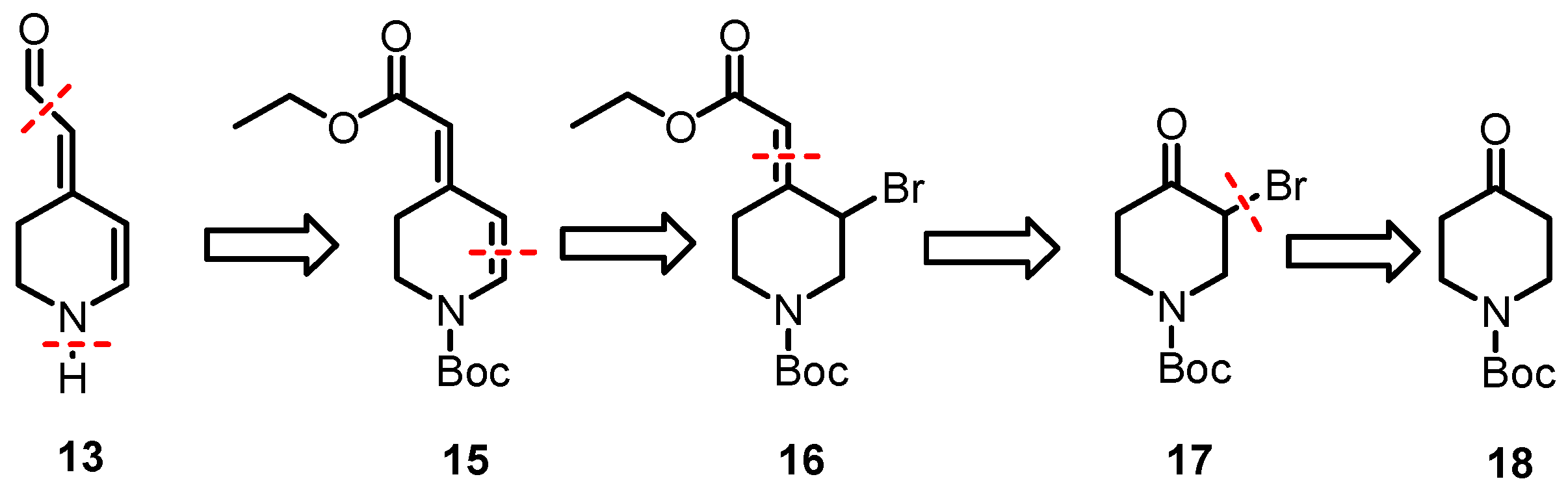
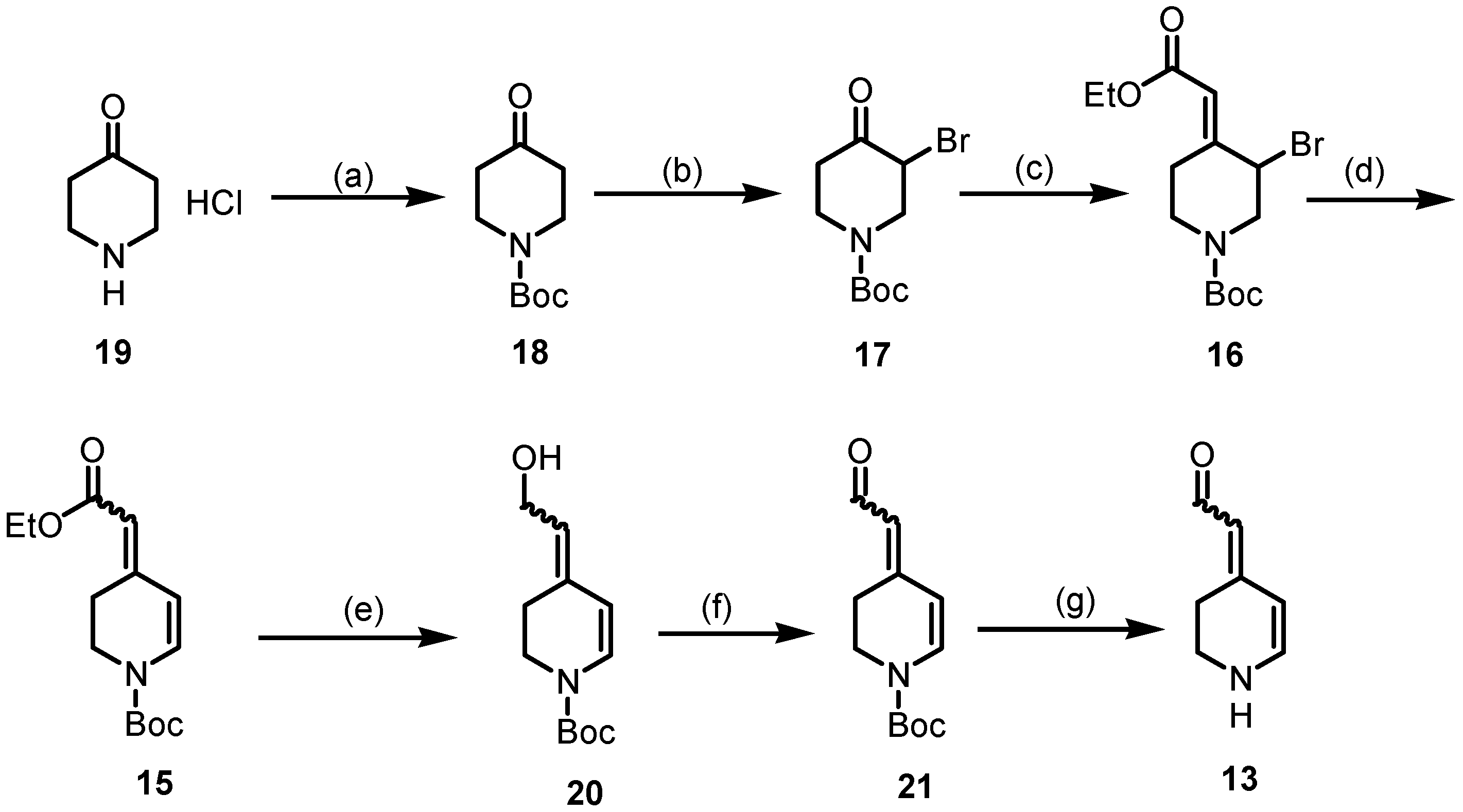
3.1.1. Synthesis of Tert-Butyl 4-Oxopiperidine-1-Carboxylate (18)
Piperidin-4-one monohydrate hydrochloride 19 (5.0 g, 32.5 mmol, 1.0 eq.) was dissolved in a 1:1 mixture of THF:H2O (100 mL) at room temperature. NaHCO3 (5.47 g, 65 mmol, 2.0 eq.) was added and the mixture was stirred for 15 min at rt. Afterwards, Boc2O (8.52 g, 39 mmol, 1.2 eq.) was added and the mixture was stirred for 16 h at room temperature. The mixture was diluted with Et2O (50 mL) and washed with aqueous solution of KHSO4 5% (3 × 50 mL), H2O (3 × 50 mL) and brine (3 × 50 mL). The combined organic layers were concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 5/5, Rf = 0.34 (cHex/EtOAc 7:3)). Colorless solid, mp 73–76 °C, yield 6.22 g (31 mmol, 96%). Exact mass (APCI): m/z = 200.1279 (calcd.200.1287 for C10H18NO3+ [M+H+]).1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.42–1.44 (m, 9H, C(CH3)3), 2.34 (t, J = 6.2 Hz, 4H, 3-(CH2)2), 3.60 (t, J = 6.2 Hz, 4H, 2-(CH2)2). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 28.0 (3C, C(CH3)3), 40.0 (2C, C-3), 40.3 (2C, C-2), 79.2 (1C, OC(CH3)3), 153.8 (1C, C(=O)OC(CH3)3), 207.4 (1C, R2C=O). FT-IR (neat): ṽ (cm−1) = 2985, 2870 (C-H, aliph.), 1724 (C=Ocarb.), 1678 (C=Oketone), 1161 (C-O).
3.1.2. Synthesis of Tert-Butyl 3-Bromo-4-Oxopiperidine-1-Carboxylate (17)
1-Boc-piperidin-4-one 18 (10 g, 50 mmol, 1.0 eq.) was dissolved in THF (30 mL) and Et2O (30 mL). AlCl3 (0.67 g, 5.0 mmol, 0.1 eq.) was added and at 0 °C, Br2 (2.6 mL, 50 mmol, 1.0 eq.) was added slowly over a period of 30 min. Afterwards, the solution was stirred at 0 °C for 18 h. Afterwards, the formed solid was filtered off and washed with Et2O. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 5/5, Rf = 0.41 (cHex/EtOAc 7:3)). Colorless solid, mp 90–93 °C, yield 6.42 g (23 mmol, 46%). Exact mass (APCI): m/z = 278.0329 (calcd.278.0392 for C10H17BrNO3+ [M+H+]). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 1.44 (s, 9H, C(CH3)3), 2.50–2.52 (m, 1H, 5-H), 2.70–2.78 (m, 1H, 5-H), 3.58–3.68 (m, 3H, 2 × 6-H, 2-H), 3.98–4.08 (m, 1H, 2-H), 4.77 (s, 1H, 3-H). 13C NMR (50 MHz, DMSO-d6): δ (ppm) = 27.9 (3C, C(CH3)3), 35.8 (1C, C-5), 42.7 (1C, C-6), 47.7 (1C, C-2), 49.0 (1C, C-3), 79.8 (1C, OC(CH3)3), 153.8 (1C, C(=O)-OC(CH3)3), 199.7 (1C, R2C=Oketone). FT-IR (neat): ṽ (cm−1) = 2978, 2931 (C-H. aliph.), 1724 (C=Oketone), 1674 (C=Ocarbamate), 1157 (C-O), 648 (C-Br).
3.1.3. Synthesis of Tert-Butyl (E)-3-Bromo-4-(Ethoxycarbonylmethylene)Piperidine-1-CarBoxylate (16)
4-(Ethoxycarbonylmethylene)triphenylphosphorane (6.9 g, 20 mmol, 1.1 eq.) was added to a solution of tert-butyl 3-bromo-4-oxopiperidine-1-carboxylate 17 (5.03 g, 18 mmol, 1.0 eq.) in CH2Cl2 (450 mL) and the reaction mixture was stirred at reflux for 2 h. Then, the mixture was cooled to room temperature and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3, Rf = 0.57 (cHex/EtOAc 7:3)). Colorless solid, mp 114–115 °C, yield 5.98 g (17 mmol, 95%). Exact mass (APCI): m/z = 348.0777 (calcd.348.0810 for C14H23BrNO4+ [M+H+]). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 1.21 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.42 (s, 9H, C(CH3)3), 2.56–2.88 (m, 2H, 5-CH2, 6-CH2), 3.34–3.51 (m, 2H, 2-CH2, 5-CH2), 4.01–4.29 (m, 4H, OCH2CH3, 6-CH2, 2-CH2), 5.04–5.12 (m, 1H, 3-CH), 6.12 (s, 1H, R2C=CH). 13C NMR (50 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 24.1 (1C, C-5), 27.9 (3C, C(CH3)3), 42.5 (1C, C-6), 51.0 (1C, C-2), 53.3 (1C, C-3), 59.9 (1C, OCH2CH3), 79.2 (1C, C(CH3)3), 113.8 (1C, R2C=CH), 153.7 (1C, C(=O)OC(CH3)3), 154.0 (1C, C-4), 165.1 (1C, CO2Et). Only one set of signals can be observed in the spectra. FT-IR (neat): ṽ (cm−1) = 2985, 2920 (C-H, aliph.), 1712 (C=O), 1670 (C=O), 1654 (C=C), 1161 (C-O), 641 (C-Br).
3.1.4. Synthesis of Tert-Butyl (E)- and (Z)-Ethoxycarbonylmethylene)-3,4-Dihydropyridine-1(2H)-Carboxylate (15)
Ester 16 (5.74 g, 16 mmol, 1.0 eq) was dissolved in dry DMF (165 mL). LiBr (8.6 g, 99 mmol, 6.0 eq.) and Li2CO3 (7.31 g, 99 mmol, 6.0 eq.) were added and the solution was stirred at 75 °C for 3 h. Then, the mixture was cooled to room temperature and extracted with EtOAc (3 × 100 mL). The combined organic layers were concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3, Rf = 0.72 (cHex/EtOAc 7:3)). Yellow oil, yield 3.88 g (14 mmol, 88%). Exact mass (APCI): m/z = 268.1497 (calcd. 268.1549 for C14H22NO4+ [M+H+]). Compound 15 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 9:1 is observed. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.46 (s, 9H, C(CH3)3), 2.49–2.53 (m, 0.2H, 5-CH2), 3.00–3.07 (m, 1.8H, 5-CH2), 3.52–3.60 (m, 2H, 6-CH2), 4.06 (q, J = 7.1 Hz, 2H, OCH2CH3), 5.32–5.34 (m, 0.1H, R2C=CH), 5.47–5.53 (m, 0.9H, 3-CH) 5.55–5.59 (m, 0.9H, R2C=CH), 6.54–6.61 (m, 0.1H, 3-CH) 7.00–7.13 (m, 0.9H, 2-CH), 7.14–7.16 (m, 0.1H, 2-CH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.2 (1C, OCH2CH3), 24.8 (0.9C, C-5), 27.7 (3C, C(CH3)3), 30.0 (0.1C, C-5), 40.0 (1C, C-6), 59.0 (1C, OCH2CH3), 81.5 (1C, C(CH3)3), 103.4 (0.1C, C-3), 108.1 (0.9C, C-3), 109.6 (0.1C, R2C=CH) 110.6 (0.9C, R2C=CH), 132.5 (0.9C, C-2), 133.0 (0.1C, C-2), 147.5 (1C, C(=O)OC(CH3)3), 147.9 (0.1C, R2C=CH) 149.1 (0.9C, R2C=CH), 166.0 (1C, CO2Et) FT-IR (neat): ṽ (cm−1) = 2978, 2931, 2900 (C-H, aliph.), 1708 (C=O), 1700 (C=O), 1608 (C=C), 1145 (C-O), 1111 (C-O).
3.1.5. Synthesis of Tert-Butyl (E)- and (Z)-4-(2-Hydroxyethylidene)-3,4-Dihydropyridine-1(2H)-Carboxylate (20)
Procedure 1
Under N2, 15 (2.80 g, 10.5 mmol, 1.0 eq.) was dissolved in dry toluene (25 mL). The solution was cooled to −78 °C, DIBAL-H (1 m solution in hexane, 31.5 mL, 31.5 mmol, 3.0 eq.) was added dropwise within 15 min, and the reaction mixture was stirred at −78 °C for 40 min. Then, at −78 °C, CH3OH (2 mL) was added carefully, and the mixture was warmed to room temperature. The mixture was filtered, the solid was washed with EtOAc (3 × 30 mL) and the combined organic layers were concentrated in vacuo. Yellow oil, yield 2.22 g (9.9 mmol, 94%).
Procedure 2
Under N2, 15 (570 mg, 2.1 mmol, 1.0 eq.) was dissolved in dry THF (5 mL) and at −10 °C, LiAlH4 (160 mg, 4.3 mmol, 2.0 eq.) was added and the mixture was stirred for 40 min. A saturated solution of potassium sodium tartrate (5 mL) was added, and the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (cHex/EtOAc 9/1 → 6/4, Rf = 0.45 (petroleum ether/EtOAc 7:3)). Yellow oil, yield 0.33 g (1.5 mmol, 70%). Exact mass (APCI): m/z = 226.1383 (calcd. 226.1443 for C12H20NO3+ [M+H+]). Compound 20 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 8.5:1.5 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.44 (s, 9H, C(CH3)3), 2.36–2.44 (m, 2H, 5-CH2), 3.48–3.55 (m, 2H, 6-CH2), 3.96–4.01 (m, 2H, CH2OH), 4.53–4.58 (m, 1H, OH), 5.13 (t, J = 6.8 Hz, 0.15H, R2C=CH), 5.35 (t, J = 6.8 Hz, 0.85H, R2C=CH), 5.40–5.46 (m, 1H, 3-CH), 6.65–6.78 (m, 1H, 2-CH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 26.9 (1C, C-5), 30.9 (3C, C(CH3)3), 43.1 (1C, C-6), 59.9 (1C, CH2OH), 83.6 (1C, C(CH3)3), 112.7 (1C, C-3), 127.8 (1C, R2C=CH), 128.4 (1C, C-2), 133.2 (1C, R2C=CH), 154.3 (1C, C(=O)OC(CH3)3). FT-IR (neat): ṽ (cm−1) = 3398 (O-H), 2974, 2931, 2873 (C-H, aliph.), 1701 (C=O), 1643 (C=C), 1612 (C=C), 1161, 1141 (C-O).
3.1.6. Synthesis of Tert-Butyl (E)- and (Z)-4-(Formylmethylene)-3,4-Dihydropyridine-1(2H)-Carboxylate (21)
CuCl (11 mg, 0.12 mmol, 0.1 eq.) and TEMPO (17 mg, 0.12 mmol, 0.1 eq.) were added to a solution of racemic mixture of allylic alcohol 20 (270 mg, 1.20 mmol, 1.0 eq.) in dry DMF (3 mL). The solution was stirred at room temperature for 16 h. Afterwards, the solution was poured into water/ice slowly and the mixture was warmed to room temperature. The solid was filtered, washed with H2O and dried. Purification by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3 Rfa = 0.38, Rfb = 0.30 (cHex/EtOAc 7:3)). Yellow oil, yield 240 mg (1.07 mmol, 90%). Exact mass (APCI): m/z = 224.1208 (calcd. 224.2495 for C24H35N2O6+ [M+H+]). Compound 21 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 6:4 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.52 (s, 3.6H, C(CH3)3), 1.53 (s, 5.4H, C(CH3)3), 2.64 (t, J = 6.8 Hz, 0.8H, 5-CH2), 2.91–3.14 (m, 1.2H, 5-CH2), 3.74 (t, J = 6.8 Hz, 2H, 6-CH2), 5.44–5.53 (m, 0.6H, 3-CH), 5.57 (d, J = 7.8 Hz, 0.4H, R2C=CH), 5.78 (d, J = 7.7 Hz, 0.6H, R2C=CH), 6.23–6.36 (m, 0.4H, 3-CH), 7.13–7.19 (m, 0.6H, 2-CH), 7.27–7.41 (m, 0.4H, 2-CH), 9.94 (d, J = 7.9 Hz, 0.6H, CHO), 10.05 (d, J = 7.9 Hz, 0.4H, CHO). 13C NMR (151 MHz, CDCl3): δ (ppm) = 24.7 (0.6C, C-5), 28.2 (3C, C(CH3)3), 30.9 (0.4C, C-5), 40.4 (1C, C-6), 82.8 (1C, C(CH3)3), 101.2 (0.4C, C-3), 108.1 (0.6C, C-3), 121.5 (1C, R2C=CH), 134.0 (0.6C, C-2), 134.5 (0.4C, C-2), 151.1 (1C, R2C=CH), 151.3 (1C, C(=O)OC(CH3)3), 189.6 (0.4C, CHO), 190.0 (0.6C, CHO). FT-IR (neat): ṽ (cm−1) = 3062, 2966, 2877 (C-H, aliph.), 1712 (C=O), 1647 (C=C), 1593 (C=C), 1141, 1122 (C-O).
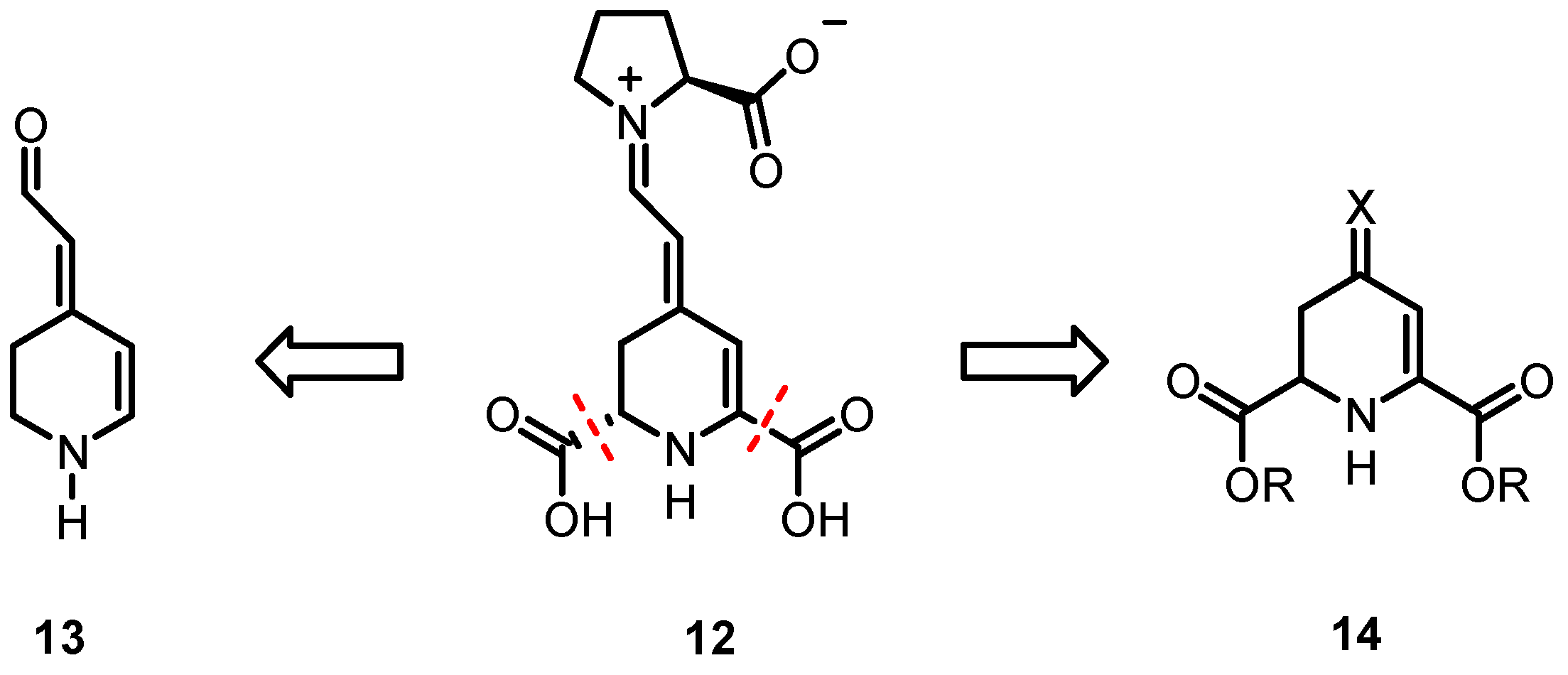
3.1.7. Synthesis of (E)- and (Z)-2-[2,3-Dihydropyridin-4(1H)-Ylidene]Acetaldehyde (13)
A solution of 21 (110 mg, 0.49 mmol, 1.0 eq.) in water (9 mL) and 1,4-dioxane (1 mL) was heated to 85 °C for 2 h. Then, the solution was cooled to room temperature and the mixture was extracted with EtOAc (6 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 1/9 Rfa = 0.15 (EtOAc)). Yellow/orange oil, yield 12 mg (0.10 mmol, 20%). The compound is highly unstable and quickly decomposed during purification. Aldehyde 13 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 6:4 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (200 MHz, CDCl3): δ (ppm) = 2.61 (t, J = 7.0 Hz, 0.8H, 5-CH2), 3.00–3.12 (m, 1.2H, 5-CH2), 3.31–3.46 (m, 2H, 6-CH2), 4.7–4.81 (m, 1H, NH), 5.19 (d, J = 7.2 Hz, 0.6H, R2C=CH), 5.29 (d, J = 7.9 Hz, 0.4H, R2C=CH), 5.60 (d, J = 8.3 Hz, 0.6H, 3-CH), 6.00 (d, J = 7.5 Hz, 0.4H, 3-CH), 6.66–6.77 (m, 1H, 2-CH), 9.79 (d, J = 8.4 Hz, 0.6H, CHO), 9.93 (d, J = 8.0 Hz, 0.4H, CHO). 13C NMR (50 MHz, CDCl3): δ (ppm) = 25.3 (0.6C, C-5), 31.75 (0.4C, C-5), 40.5 (0.6C, C-6), 40.9 (0.4C, C-6), 93.9 (0.4C, C-3), 100.3 (0.6C, C-3), 116.2 (0.4C, R2C=CH), 117.1 (0.6C, R2C=CH), 142.6 (1C, C-2), 143.4 (1C, C-2), 155.3 (0.4C, R2C=CH), 171.3 (0.6C, R2C=CH), 189.2 (0.4C, CHO), 189.3 (0.6C, CHO).
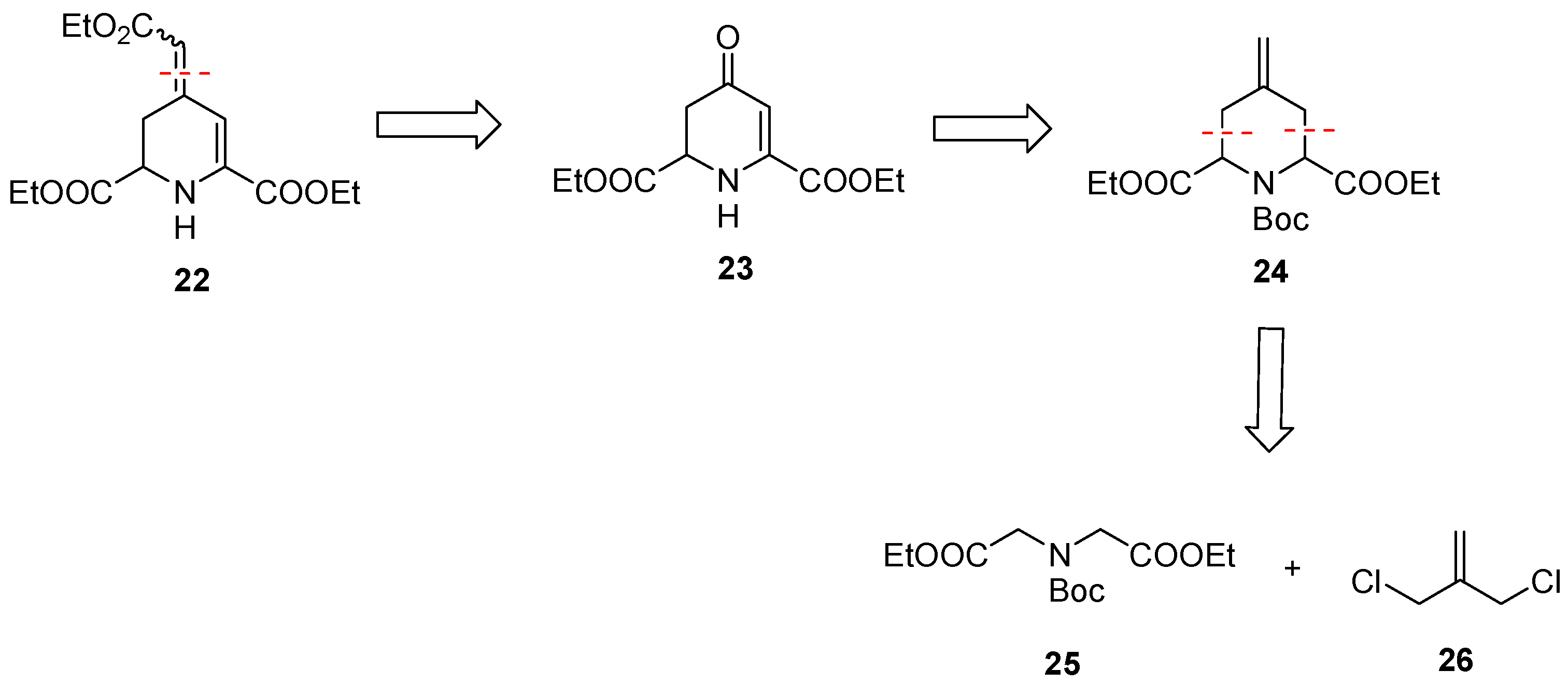
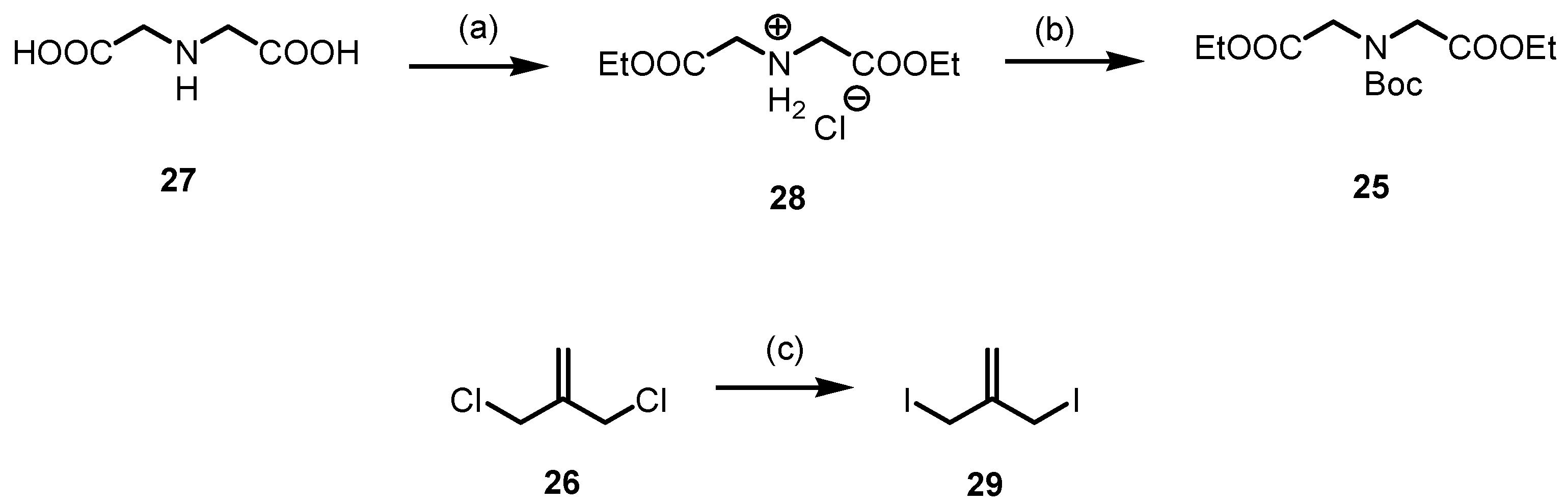
3.1.8. Synthesis of Diethyl 2,2′-Iminodiacetate∙HCl (28)
At 0 °C, SOCl2 (20.0 mL, 275 mmol, 1.5 eq) was added dropwise to a suspension of iminodiacetic acid 27 (24.4 g, 184 mmol, 1.0 eq) in EtOH abs. (200 mL). Afterwards, the reaction mixture was heated to reflux for 16 h. The solution was cooled down to room temperature and concentrated in vacuo. Colorless solid, mp 88–89 °C, yield 40.6 g (98%). C8H16ClNO4 (225.7 g/mol). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.24 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 3.70 (brs, 2H, NH2+), 3.99 (s, 4H, 2 × CH2), 4.21 (q, J = 7.1 Hz, 4H, 2 × OCH2CH3). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 13.9 (2C, 2 × OCH2CH3), 46.4 (2C, 2 × CH2), 61.8 (2C, 2 × OCH2CH3), 166.4 (2C, 2 × O=COEt). IR (neat): ṽ (cm−1) = 2936 (C-Halip), 1736 (C=Oester), 1204, 1076, 1015 (C-N, C-O). Exact mass (APCI): m/z = 190.1073 (calcd. 190.1074 for C8H16NO4 [M-Cl]+).
3.1.9. Synthesis of Diethyl 2,2′-[N-(Tert-Butoxycarboxyl)Imino]Diacetate (25)
NaHCO3 (22.8 g, 271 mmol, 3.0 eq) was added to a solution of 28 (20.4 g, 90.4 mmol, 1.0 eq) in THF (80 mL) and H2O (20 mL). The reaction mixture was stirred for 30 min at room temperature. After the addition of Boc2O (19.3 mL, 90.4 mmol, 1.0 eq), the mixture was stirred at room temperature for 16 h. Then, it was extracted with EtOAc (3 × 50 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 8 cm, h = 16 cm, cHex/EtOAc 8:2, V = 80 mL). Colorless oil, yield 19.9 g (76%). C13H23NO6 (289.3 g/mol). TLC: Rf = 0.36 (cHex/EtOAc 8:2). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.20 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.35 (s, 9H, C(CH3)3), 3.98 (s, 2H, NCH2), 4.01 (s, 2H, NCH2), 4.10 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3). Ratio of rotamers is 1:1. 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 14.1 (1C, OCH2CH3), 27.7 (3C, C(CH3)3), 49.2 (1C, CH2), 49.7 (1C, CH2), 60.4 (2C, OCH2CH3), 79.9 (1C, C(CH3)3), 154.6 (1C, N(C=O)O), 169.45 (1C, O=COEt), 169.53 (1C, O=COEt). Ratio of rotamers is 1:1. IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1747 (C=Oester), 1700 (C=Ocarbamate), 1185, 1159, 1026 (C-N, C-O). Exact mass (APCI): m/z = 290.1587 (calcd. 290.1598 for C13H24NO6+ [M+H]+).
3.1.10. Synthesis of 3-Iodo-2-(Iodomethyl)Prop-1-Ene (29) [44]
NaI (17.8 g, 119 mmol, 2.5 eq) was added to a solution of 3-chloro-2-(chloromethyl)prop-1-ene 26 (5.50 mL, 47.5 mmol, 1.0 eq) in acetone (100 mL) and the mixture was stirred at reflux for 16 h. The suspension was cooled to room temperature and concentrated in vacuo. The residue was dissolved in H2O (75 mL) and cHex (75 mL). After separation of the two layers, the organic layer was washed with Na2SO3 (2 × 50 mL) and H2O (50 mL), dried (Na2SO4), filtered and concentrated in vacuo. Light green solid, mp 28–29 °C, yield 14.5 g (99%). C4H6I2 (307.9 g/mol). 1H NMR (600 MHz, CD3OD): δ (ppm) = 4.21 (s, 4H, 2 × CH2I), 5.40 (s, 2H, R2C=CH2). 13C NMR (151 MHz, CD3OD): δ (ppm) = 6.6 (2C, 2 × CH2I), 116.4 (1C, R2C=CH2), 146.0 (1C, R2C=CH2). Exact mass (APCI): m/z = 308.8637 (calcd. 308.8632 for C4H7I2+ [M+H]+).
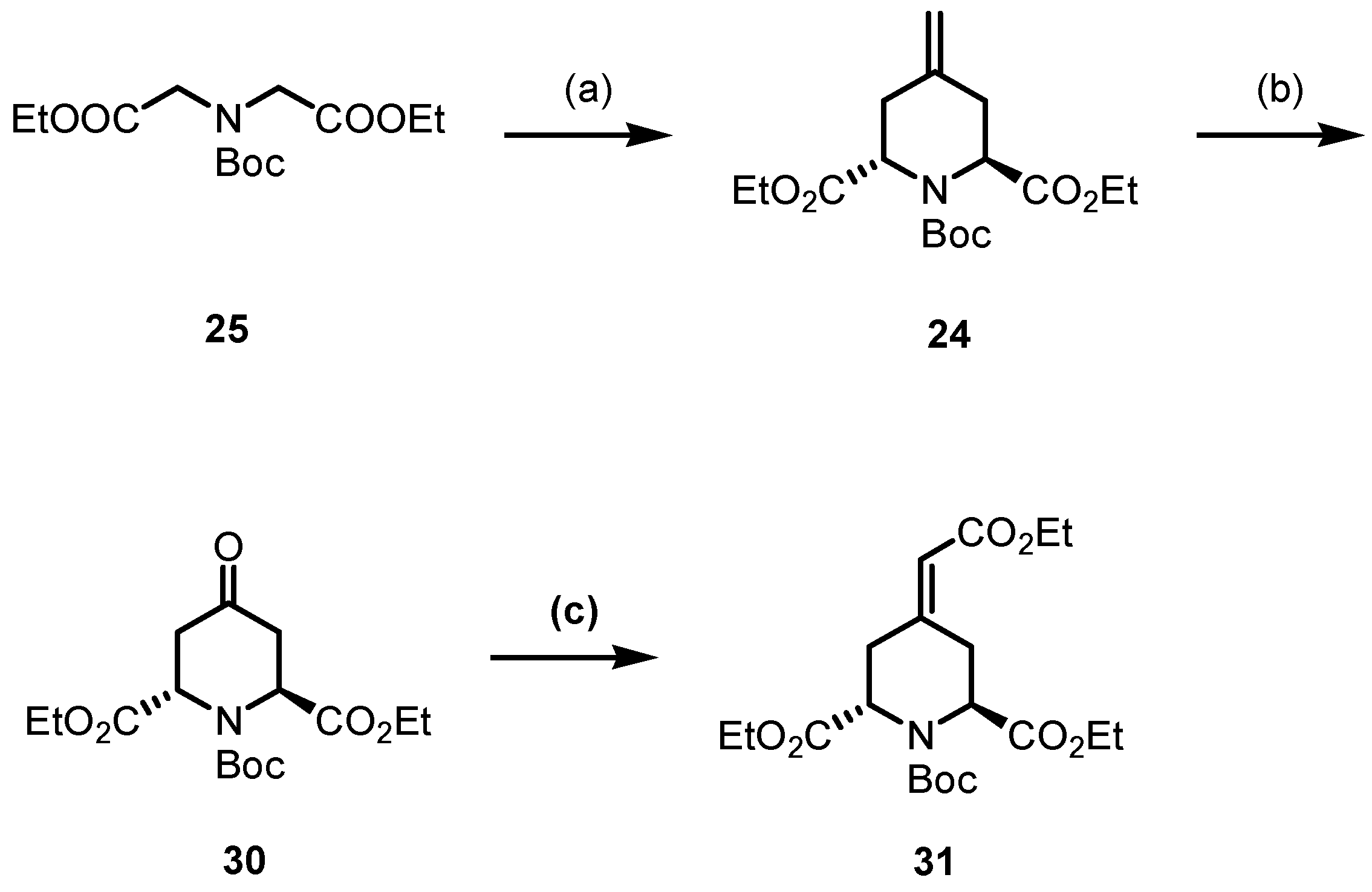
3.1.11. Synthesis of 1-Tert-Butyl 2,6-Diethyl Cis- and Trans-4-Methylenepiperidine-1,2,6-Tricarboxylate (24)
At −78 °C, n-BuLi (1.6 m in n-hexane, 74.4 mL, 119 mmol, 2.1 eq) was added dropwise to a solution of i-Pr2NH (16.7 mL, 119 mmol, 2.1 eq) in dry THF (170 mL). After the mixture was stirred for 1 h, a solution of 25 (16.4 g, 56.7 mmol, 1.0 eq) in dry THF (15 mL) was added and the mixture was stirred for 1 h at −78 °C. Then, a solution of 29 (22.1 g, 68.1 mmol, 1.2 eq) in dry THF (15 mL) was added and the reaction mixture was stirred for 30 min at −78 °C and warmed up to room temperature over 16 h. At 0 °C, H2O (150 mL) was added and the mixture was extracted with EtOAc (3 × 80 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified twice by flash column chromatography (1. ø = 8 cm, h = 25 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL, 2. ø = 8 cm, h = 25 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL). Yellow oil, yield 14.8 g (77%). C17H27NO6 (341.4 g/mol). TLC: Rf = 0.26 (cHex/EtOAc 8:2). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.42 (s, 9 × 0.9H, C(CH3)3), 1.47 (s, 9 × 0.1H, C(CH3)3*), 2.43–2.51 (m, 2 × 0.1H, 3/5-CH2*), 2.65 (dd, J = 15.9/2.7 Hz, 1H, 3/5-CH2), 2.74 (dd, J = 16.0/3.6 Hz, 1H, 3/5-CH2), 2.79–2.87 (m, 2 × 0.9H, 3/5-CH2), 4.09–4.24 (m, 4H, 2 × OCH2CH3), 4.60 (dd, J = 3.5/3.0 Hz, 1H, 2/6-CH), 4.69 (dd, J = 6.2/4.1 Hz, 1H, 2/6-CH), 4.81–4.83 (m, 2 × 0.9H, R2C=CH2), 4.90–4.93 (m, 2 × 0.1H, R2C=CH2*). Ratio of isomers is 9:1 (trans:cis). Signals for the cis diastereomer are marked with an asterisk (*). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (1C, OCH2CH3), 14.5 (1C, OCH2CH3), 28.3 (3 × 0.9C, C(CH3)3), 28.4 (3 × 0.1C, C(CH3)3*), 33.25 (0.9C, C-3/5), 33.34 (0.9C, C-3/5), 33.6 (2 × 0.1C, C-3*, C-5*), 54.6 (1C, C-2/6), 55.7 (1C, C-2/6), 61.0 (2 × 0.1C, OCH2CH3*), 61.2 (0.9C, OCH2CH3), 61.3 (0.9C, OCH2CH3), 81.17 (0.9C, C(CH3)3), 81.21 (0.1C, C(CH3)3*) 112.3 (0.9C, R2C=CH2), 112.7 (0.1C, R2C=CH2*), 137.5 (0.9C, R2C=CH2), 138.1 (0.1C, R2C=CH2*), 155.2 (0.1C, N(C=O)O*), 155.5 (0.9C, N(C=O)O), 171.3 (2 × 0.1C, O=COEt)*), 172.8 (2 × 0.9C, O=COEt). Ratio of isomers is 9:1 (trans:cis). Signals for the cis diastereomer are marked with an asterisk (*). Purity (HPLC, method A): 94.4% (tR = 21.5 min). IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1740 (C=Oester), 1701 (C=Ocarbamate), 1655 (C=C), 1180, 1165, 1022 (C-N, C-O). Exact mass (APCI): m/z = 342.1913 (calcd. 342.1911 for C17H28NO6 [M+H]+).
3.1.12. Synthesis of 1-Tert-Butyl 2,6-Diethyl Trans-4-Oxopiperidine-1,2,6-Tricarboxylate (30)
OsO4 (0.05 m in H2SO4, 3.60 mL, 0.18 mmol, 0.01 eq), pyridine (0.70 mL, 9.10 mmol, 0.5 eq) and NaIO4 (15.6 g, 72.8 mmol, 4.0 eq) were added to a solution of 24 (6.20 g, 18.2 mmol, 1.0 eq) in t-BuOH (80 mL) and H2O (120 mL) and the suspension was stirred at room temperature for 48 h. Then, the mixture was filtered and Na2SO3 (50 mL) and EtOAc (50 mL) were added to the solution. The aqueous layer was extracted with EtOAc (3 × 50 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 8 cm, h = 16 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL). Colorless solid, mp 49–50 °C, yield 4.55 g (73%). C16H25NO7 (343.4 g/mol). TLC: Rf = 0.33 (cHex/EtOAc 8:2). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.45 (s, 9H, C(CH3)3), 2.72 (dd, J = 18.0/1.4 Hz, 1H, 3/5-CHeq), 2.86 (dd, J = 18.0/1.4 Hz, 1H, 3/5-CHeq), 3.01–3.08 (m, 2H, 3-CHax, 5-CHax), 4.12–4.26 (m, 4H, 2 × OCH2CH3), 4.83 (d, J = 7.8 Hz, 1H, 2/6-CH), 5.06 (d, J = 7.8 Hz, 1H, 2/6-CH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.2 (1C, OCH2CH3), 14.3 (1C, OCH2CH3), 28.3 (3C, C(CH3)3), 40.6 (1C, C-3/5), 41.0 (1C, C-3/5), 53.0 (1C, C-2/6), 54.2 (1C, C-2/6), 61.9 (1C, OCH2CH3), 62.1 (1C, OCH2CH3), 81.9 (1C, C(CH3)3), 154.5 (1C, N(C=O)O), 172.3 (1C, O=COEt), 172.4 (1C,O=COEt), 203.8 (1C, C-4). IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1736 (C=Oester), 1701 (C=Oketone, carbamate), 1188, 1115, 1026 (C-N, C-O). Exact mass (APCI): m/z = 344.1690 (calcd. 344.1704 for C16H26NO7+ [M+H]+).
3.1.13. Synthesis of 1-Tert-Butyl 2,6-Diethyl Trans-(Ethoxycarbonylmethylene)-Piperidine-1,2,6-Tricarboxylate (31)
4-(Ethoxycarbonylmethylene)triphenylphosphorane (7.60 g, 21.7 mmol, 1.75 eq) was added to a solution of 30 (4.30 g, 12.4 mmol, 1.0 eq) in toluene (50 mL) and the mixture was heated at reflux for 48 h. After concentrating the mixture in vacuo, the residue was purified by flash column chromatography (ø = 6 cm, h = 17 cm, cHex/EtOAc 9:1, V = 80 mL). Colorless oil, yield 3.9 g (75%). C20H31NO8 (413.5 g/mol). TLC: Rf = 0.35 (cHex/EtOAc 8:2). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.11–1.22 (m, 9H, 3 × OCH2CH3), 1.35 (s, 9 × 0.54H, C(CH3)3), 1.36 (s, 9 × 0.46H, C(CH3)3*), 2.71 (dd, J = 16.8/2.2 Hz, 0.54H, 3/5-CH2), 2.78 (dd, J = 17.1/3.2 Hz, 0.46H, 3/5-CH2*), 2.83–2.95 (m, 1.46H, 3/5-CH2, 3-CH2*, 5-CH2*), 3.01 (ddm, J = 18.9/7.3 Hz, 0.54H, 3/5-CH2), 3.61 (dm, J = 18.9 Hz, 0.54H, 3/5-CH2), 3.68 (dm, J = 18.5 Hz, 0.46H, 3/5-CH2*), 4.02–4.17 (m, 6H, 3 × OCH2CH3), 4.57 (dd, J = 6.7/2.3 Hz, 0.54H, 2/6-CH), 4.62 (dd, J = 6.4/3.1 Hz, 0.46H, 2/6-CH*), 4.67 (dd, J = 7.2/1.9 Hz, 0.46H, 2/6-CH*), 4.75 (dd, J = 7.3/2.3 Hz, 0.54H, 2/6-CH), 5.80 (s, 0.54H, R2C=CH), 5.81 (s, 0.46H, R2C=CH*). Ratio of rotamers is 54:46. Signals for the minor rotamer are marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 13.9 (0.54C, OCH2CH3), 14.0 (0.46C, OCH2CH3*), 14.0 (0.46C, OCH2CH3*), 14.1 (0.54C, OCH2CH3), 14.11 (1C, OCH2CH3), 27.7 (s, 3C, C(CH3)3), 29.8 (0.46C, C-3/5*), 30.4 (0.54C, C-3/5), 33.8 (0.54C, C-3/5), 34.0 (0.46C, C-3/5*), 51.6 (0.54C, C-2/6), 53.0 (0.46C, C-2/6*), 53.1 (0.46C, C-2/6*), 54.1 (0.54C, C-2/6), 59.5 (1C, OCH2CH3), 60.8 (0.46C, OCH2CH3*), 60.9 (0.54C, OCH2CH3), 61.0 (0.46C, OCH2CH3*), 61.1 (0.54C, OCH2CH3), 80.3 (0.54C, C(CH3)3), 80.4 (0.46C, C(CH3)3*), 116.9 (0.54C, R2C=CH2), 117.0 (0.46C, R2C=CH2*), 151.4 (0.46C, R2C=CH2*), 151.6 (0.54C, R2C=CH2), 154.0 (0.54C, N(C=O)O), 154.2 (0.46C, N(C=O)O*), 165.0 (1C, O=COEt), 171.5 (0.46C, O=COEt*), 171.7 (0.54C, O=COEt), 172.0 (0.54C, O=COEt), 172.2 (0.46C, O=COEt*). Ratio of rotamers is 54:46. Signals for the minor rotamer are marked with an asterisk (*). Purity (HPLC, method A): 98.9% (tR = 22.1 min). IR (neat): ṽ (cm−1) = 2978 (C-Haliph), 1744 (C=Oester), 1701 (C=Ocarbamate), 1651 (C=C), 1184, 1142, 1026 (C-N, C-O). Exact mass (APCI): m/z = 414.2144 (calcd. 414.2122 for C20H32NO8+ [M+H]+).
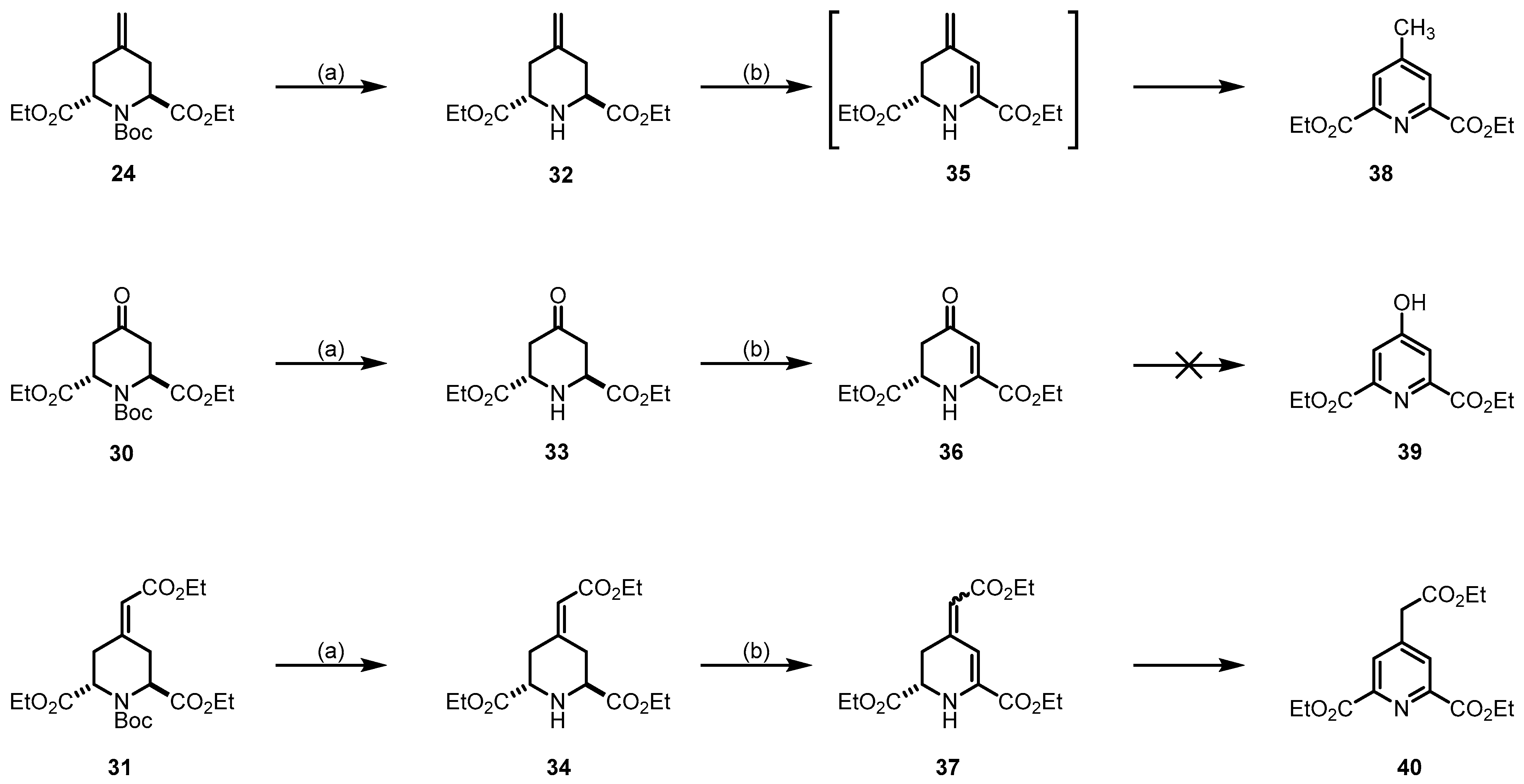
3.1.14. Synthesis of Diethyl Cis/Trans-4-Methylenepiperidine-2,6-Dicarboxylate (32)
TFA (6.5 mL, 87.9 mmol, 30 eq) was added to a solution of 24 (1.0 g, 2.9 mmol, 1.0 eq) in dry CH2Cl2 (50 mL) and the mixture was stirred at room temperature for 16 h. The next day, Na2CO3 was added, the layers were separated and the aqueous layer was extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The diastereomers were separated twice by flash column chromatography (1.50 g cartridge, cHex/EtOAc 8:2 → 6:4; 2. 25 g cartridge, cHex/EtOAc 8:2). C12H19NO4 (241.3 g/mol). cis-32: Yellow oil, yield 79 mg (11%). TLC: Rf = 0.31 (cHex/EtOAc 1:1). 1H NMR (600 MHz CDCl3) δ = 1.29 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.10–2.17 (m, 2H, 3-CHax, 5-CHax), 2.62 (dd, J = 13.5/2.7 Hz, 2H, 3-CHeq, 5-CHeq), 3.37 (dd, J = 11.8/3.0 Hz, 2H, 2-CHax, 6-CHax), 4.22 (dq, J = 7.1/1.4 Hz, 4H, 2 × OCH2CH3), 4.87 (t, J = 1.7 Hz, 2H, R2C=CH2). NH signal is missing. 13C NMR (151 MHz, CDCl3) δ = 14.3 (2C, 2 × OCH2CH3), 37.7 (2C, C-3, C-5), 58.9 (2C, C-2, C-6), 61.4 (2C, 2 × OCH2CH3), 111.4 (1C, R2C=CH2), 142.5 (1C, C-4), 171.9 (2C, 2 × O=COEt)). IR (neat): ṽ (cm−1) = 2982 (C-Haliph), 1732 (C=Oester), 1651 (C=C), 1180, 1026 (C-N, C-O). Exact mass (APCI): m/z = 242.1360 (calcd. 242.1387 for C12H20NO4 [M+H]+). trans-32: Yellow oil, yield 623 mg (89%). TLC: Rf = 0.20 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.29 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.46 (dd, J = 13.2/7.0 Hz, 2H, 3-CH2, 5-CH2), 2.56 (dd, J = 13.2/5.0 Hz, 2H, 3-CH2, 5-CH2), 3.84 (dd, J = 7.0/5.0 Hz, 2H, 2-CH, 6-CH), 4.14–4.24 (m, 4H, 2 × OCH2CH3), 4.87 (s, 2H, R2C=CH2). NH signal is missing. 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.4 (2C, 2 × OCH2CH3), 36.3 (2C, C-3, C-5), 56.1 (2C, C-2, C-6), 61.2 (2C, 2 × OCH2CH3), 111.7 (1C, R2C=CH2), 141.3 (1C, C-4), 172.7 (2C, 2 × O=COEt). IR (neat): ṽ (cm−1) = 3356 (N-H), 2978 (C-Halip), 1728 (C=Oester), 1655 (C=C), 1200, 1165, 1026 (C-N, C-O). Exact mass (APCI): m/z = 242.1373 (calcd. 242.1387 for C12H20NO4+ [M+H]+).
3.1.15. Synthesis of Diethyl Trans-4-Oxopiperidine-2,6-Dicarboxylate (33)
TFA (3.60 mL, 47.0 mmol, 30 eq) was added to a solution of 30 (0.54 g, 1.57 mmol, 1 eq) in CH2Cl2 (15 mL) and the mixture was stirred at room temperature for 16 h. Then, the mixture was washed with NaHCO3 (30 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Yellow oil, yield 0.27 g (71%). C11H17NO5 (243.3 g/mol). TLC: Rf = 0.22 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.61 (ddd, J = 15.1/7.0/1.4 Hz, 2H, 3-CHax, 5-CHax), 2.71 (ddd, J = 15.0/5.5/1.3 Hz, 2H, 3-CHeq, 5-CHeq), 4.05 (dd, J = 7.0/5.5 Hz, 2H, 2-CH, 6-CH), 4.21 (q, J = 7.1 Hz, 4H, 2 × OCH2CH3). The signal for NH is not observed in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (2C, 2 × OCH2CH3), 42.7 (2C, C-3, C-5), 54.8 (2C, C-2, C-6), 61.8 (2C, 2 × OCH2CH3), 171.7 (2C, 2 × O=COEt), 204.7 (1C, C-4).
3.1.16. Synthesis of Diethyl Trans-4-(2-Ethoxy-2-Oxoethylidene)Piperidine-2,6-Dicarboxylate (34)
TFA (2.10 mL, 28.4 mmol, 30 eq) was added to a solution of 31 (0.39 g, 0.95 mmol, 1 eq) in CH2Cl2 (10 mL) and the mixture was stirred at room temperature for 16 h. Then, the mixture was washed with NaHCO3 (20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 15 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Yellow oil, yield 0.27 g (90%). C15H23NO6 (313.4 g/mol). TLC: Rf = 0.35 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.52 (dd, J = 13.3/7.1 Hz, 1H, 3/5-CHax), 2.61 (dd, J = 13.3,/5.0 Hz, 1H, 3/5-CHeq), 3.21 (dd, J = 13.8, 6.9 Hz, 1H, 3/5-CHax), 3.26 (dd, J = 13.8/5.3 Hz, 1H, 3/5-CHeq), 3.86 (dd, J = 6.9/5.3 Hz, 1H, 2/6-CH), 3.92 (dd, J = 7.1/5.0 Hz, 1H, 2/6-CH), 4.12–4.24 (m, 6H, 3 × OCH2CH3), 5.75 (s, 1H, R2C=CH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (1C, OCH2CH3), 14.4 (2C, 2 × OCH2CH3), 31.2 (1C, C-3/5), 38.2 (1C, C-3/5), 55.6 (1C, C-2/6), 56.0 (1C, C-2/6), 60.0 (1C, OCH2CH3), 61.3 (1C, OCH2CH3), 61.4 (1C, OCH2CH3), 117.5 (1C, R2C=CH), 153.4 (1C, R2C=CH), 166.0 (1C, O=COEt), 172.2 (1C, O=COEt), 172.6. (1C, O=COEt).
3.1.17. Synthesis of Diethyl 4-Methylpyridine-2,6-Dicarboxylate (38)
At 0 °C, NaOCl (0.75 m in H2O, 3.90 mL, 2.90 mmol, 2 eq) was added to a solution of 32 (350 mg, 1.45 mmol, 1 eq) and AcOH (0.17 mL, 2.90 mmol, 2 eq) in t-BuOH (0.20 mL, 1.74 mmol, 1.2 eq) and methyl t-butyl ether (8 mL) and the mixture was stirred for 1 h. Then, DIPEA (2.05 mL, 11.6 mmol, 8 eq) was added and the mixture was stirred at room temperature for 16 h. After the addition of H2O (10 mL), the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 95:5 → 9:1). Yellow oil, yield 20 mg (6%). C12H15NO4 (237.3 g/mol). TLC: Rf = 0.59 (cHex/EtOAc 9:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.45 (t, J = 7.2 Hz, 6H, 2 × OCH2CH3), 2.51 (s, 3H, CH3), 4.47 (q, J = Hz, 4H, 2 × OCH2CH3), 8.10 (s, 2H, 3-CHarom, 5-CHarom). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.4 (2C, 2 × OCH2CH3), 21.3 (1C, CH3), 62.4 (2C, 2 × OCH2CH3), 128.8 (2C, C-3arom, C-5arom), 148.6 (2C, C-2arom, C-6arom), 150.2 (1C, C-4arom), 165.1 (2C, 2 × O=COEt).
3.1.18. Synthesis of Diethyl (RS)-4-Oxo-1,2,3,4-Tetrahydropyridine-2,6-Dicarboxylate (36)
At 0 °C, NaOCl (0.75 m in H2O, 1.70 mL, 1.28 mmol, 1.2 eq) was added to a solution of 33 (260 mg, 1.07 mmol, 1 eq) and AcOH (0.07 mL, 1.28 mmol, 1.2 eq) in t-BuOH (0.12 mL, 1.28 mmol, 1.2 eq) and methyl t-butyl ether (10 mL) and the mixture was stirred for 1 h. Then, DIPEA (0.90 mL, 5.35 mmol, 5 eq) was added and the mixture was stirred at room temperature for 16 h. H2O (15 mL) was added and the mixture was extracted with EtOAc (3 × 15 mL) and dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 7:3 → 1:1). Colorless oil, yield 101 mg (39%). C11H15NO5 (241.2 g/mol). TLC: Rf = 0.40 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.35 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.70 (dd, J = 16.5/12.3 Hz, 1H, 3-CH2), 2.80 (dd J = 16.5/5.8 Hz, 1H, 3-CH2), 4.23–4.29 (m, 2H, OCH2CH3), 4.32–4.37 (m, 3H, 2-CH, OCH2CH3), 5.77 (s, 1H, 5-CH), 6.07 (s, 1H, NH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.1 (1C, OCH2CH3), 14.2 (1C, OCH2CH3), 38.2 (1C, C-3), 54.8 (1C, C-2), 62.4 (1C, OCH2CH3), 63.0 (1C, OCH2CH3), 102.2 (1C, C-5), 147.7 (1C, C-6), 162.9 (1C, O=COEt), 169.9 (1C, O=COEt), 192.5 (1C, C-4).
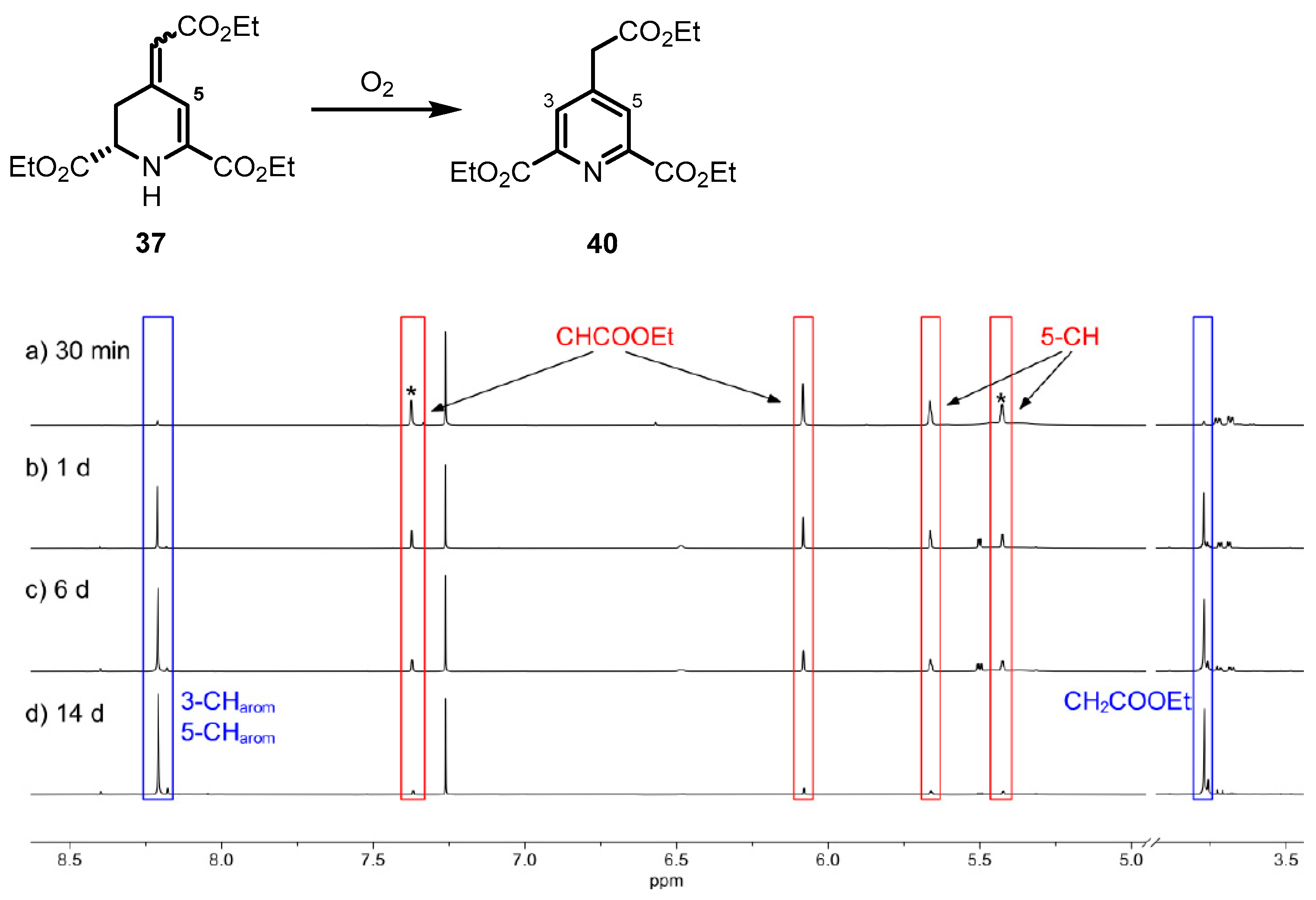
3.1.19. Synthesis of Diethyl (E)- and (Z)-(RS)-4-(2-Ethoxy-2-Oxoethylidene)-1,2,3,4-Tetrahydropyridine-2,6-Dicarboxylate (37)
At 0 °C, NaOCl (0.75 m in H2O, 2.20 mL, 1.65 mmol, 2 eq) was added to a solution of 34 (260 mg, 0.82 mmol, 1 eq) and AcOH (0.10 mL, 1.65 mmol, 2 eq) in t-BuOH (0.10 mL, 1.00 mmol, 1.2 eq) and methyl t-butyl ether (8 mL) and the mixture was stirred for 1 h. Then, DIPEA (1.15 mL, 6.61 mmol, 8 eq) was added and the mixture was stirred at room temperature for 16 h. H2O (10 mL) was added and the mixture was extracted with EtOAc (3 × 15 mL) and dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 8:2 → 6:4). Yellow oil, yield 77 mg (30%). C15H21NO6 (311.3 g/mol). TLC: Rf = 0.49 (cHex/EtOAc 7:3). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.24–1.32 (m, 6H, 2 × OCH2CH3, 2 × OCH2CH3*), 1.34 (t, J = 7.1 Hz, 3 × 0.55H, OCH2CH3), 1.35 (t, J = 7.1 Hz, 3 × 0.45H, OCH2CH2*), 2.76 (ddd, J = 15.5/9.6/1.5 Hz, 0.45H, 3-CH2*), 2.83 (ddd, J = 15.5/5.0/1.2 Hz, 0.45H, 3-CH2*), 3.10 (ddd, J = 17.0/10.2/2.0 Hz, 0.55H, 3-CH2), 3.70 (ddd, J = 17.0/4.8/1.6 Hz, 0.55H, 3-CH2), 4.00 (dd, J = 10.2/4.8 Hz, 0.55H, 2-CH), 4.06 (dd, J = 9.6/5.0 Hz, 0.45H, 2-CH*), 4.12–4.27 (m, 4H, 2 × OCH2CH3, 2 × OCH2CH3*), 4.30 (q, J = 7.1 Hz, 1.10H, OCH2CH3), 4.31 (q, J = 7.1 Hz, 0.9H, OCH2CH3*), 5.43 (s, 0.45H, 5-CH*), 5.66 (s, 0.55H, 5-CH), 6.08 (s, 0.55H, CHCOOEt), 7.37 (s, 0.45H, CHCOOEt*). Ratio of diastereomers is 55:45. The signals for the minor diastereomer are marked with an asterisk (*). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.25, 14.28, 14.20, 14.31 (2C, 2 × OCH2CH3, 2 × OCH2CH3*), 14.50, 14.51 (1C, OCH2CH3, OCH2CH3*), 28.4 (0.55C, 3-C), 33.7 (0.45C, C-3*), 53.5 (0.55C, C-2), 55.8 (0.45C, C-2*), 59.82 (0.55, OCH2CH3), 59.84 (0.45C, OCH2CH3*), 61.8 (0.45C, OCH2CH3*), 61.9 (0.55C, OCH2CH3), 62.06 (0.55C, OCH2CH3), 62.13 (0.45C, OCH2CH3*), 102.4 (0.45C, CHCOOEt*), 107.1 (0.55C, CHCOOEt), 112.3 (0.45C, C-5*), 113.2 (0.55C, C-5), 137.9 (0.55C, C-6), 138.1 (0.45C, C-6*), 146.1 (0.45C, C-4*), 148.0 (0.55C, C-4), 163.2 (0.55C, O=COEt), 163.9 (0.45C, O=COEt*), 166.7 (0.45C, O=COEt*), 167.0 (0.55C, O=COEt), 170.8 (0.45C, O=COEt*), 171.2 (0.55C, O=COEt). Ratio of diastereomers is 55:45. The signals for the minor diastereomer are marked with an asterisk (*).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











