

Targeting SARS-CoV-2 nsp13 Helicase and Assessment of Druggability Pockets: Identification of Two Potent Inhibitors by a Multi-Site In Silico Drug Repurposing Approach
 , ,
, ,  ,
,  , , , , ,
, , , , ,
Abstract
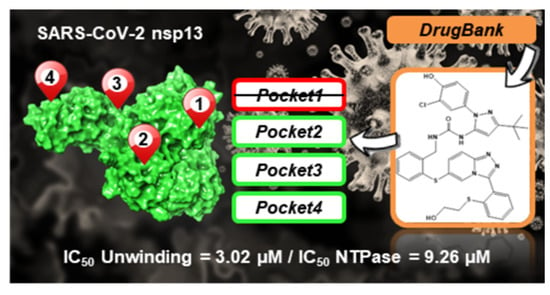
:
1. Introduction
2. Results
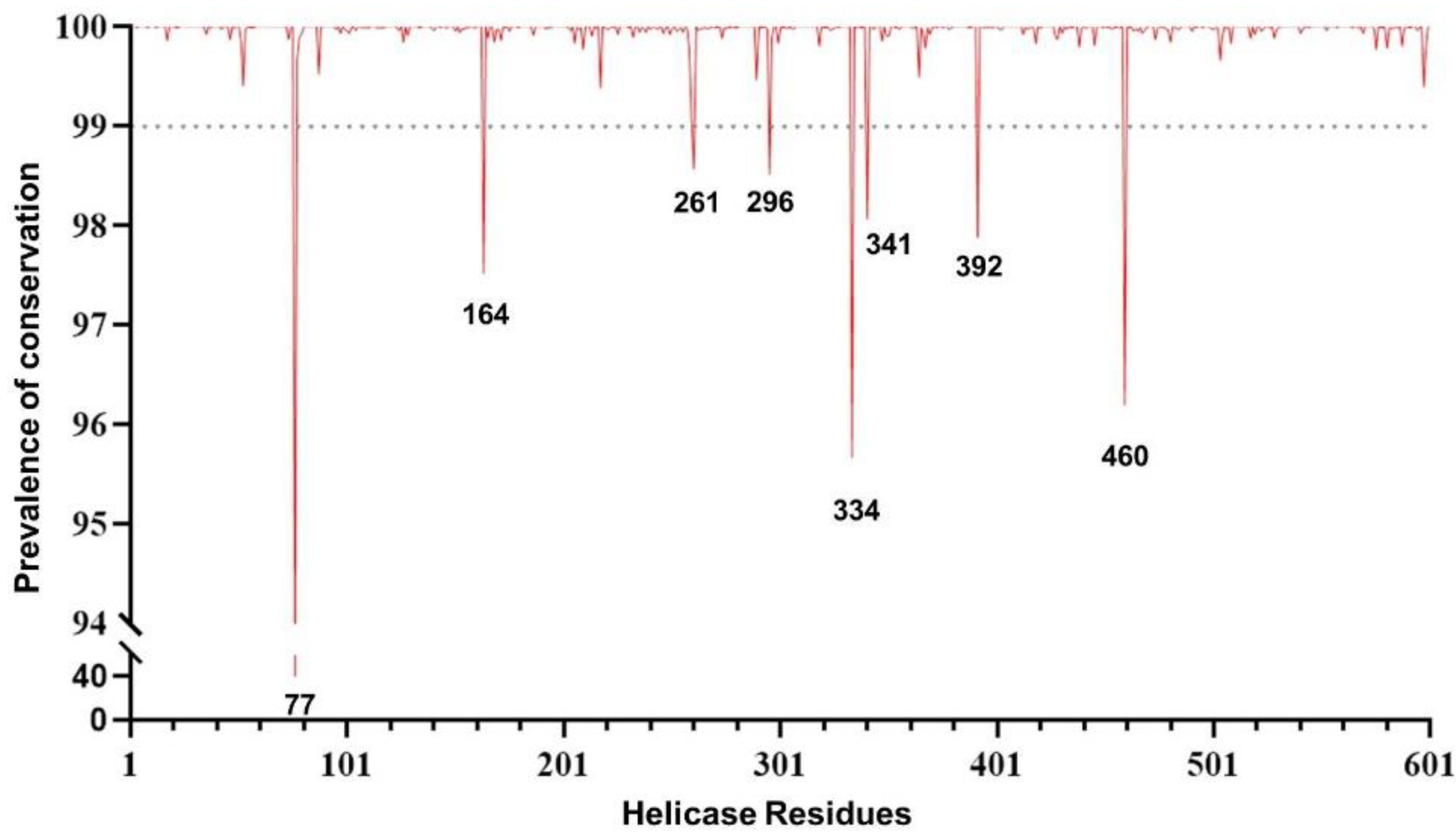
2.1. Conservation Analysis
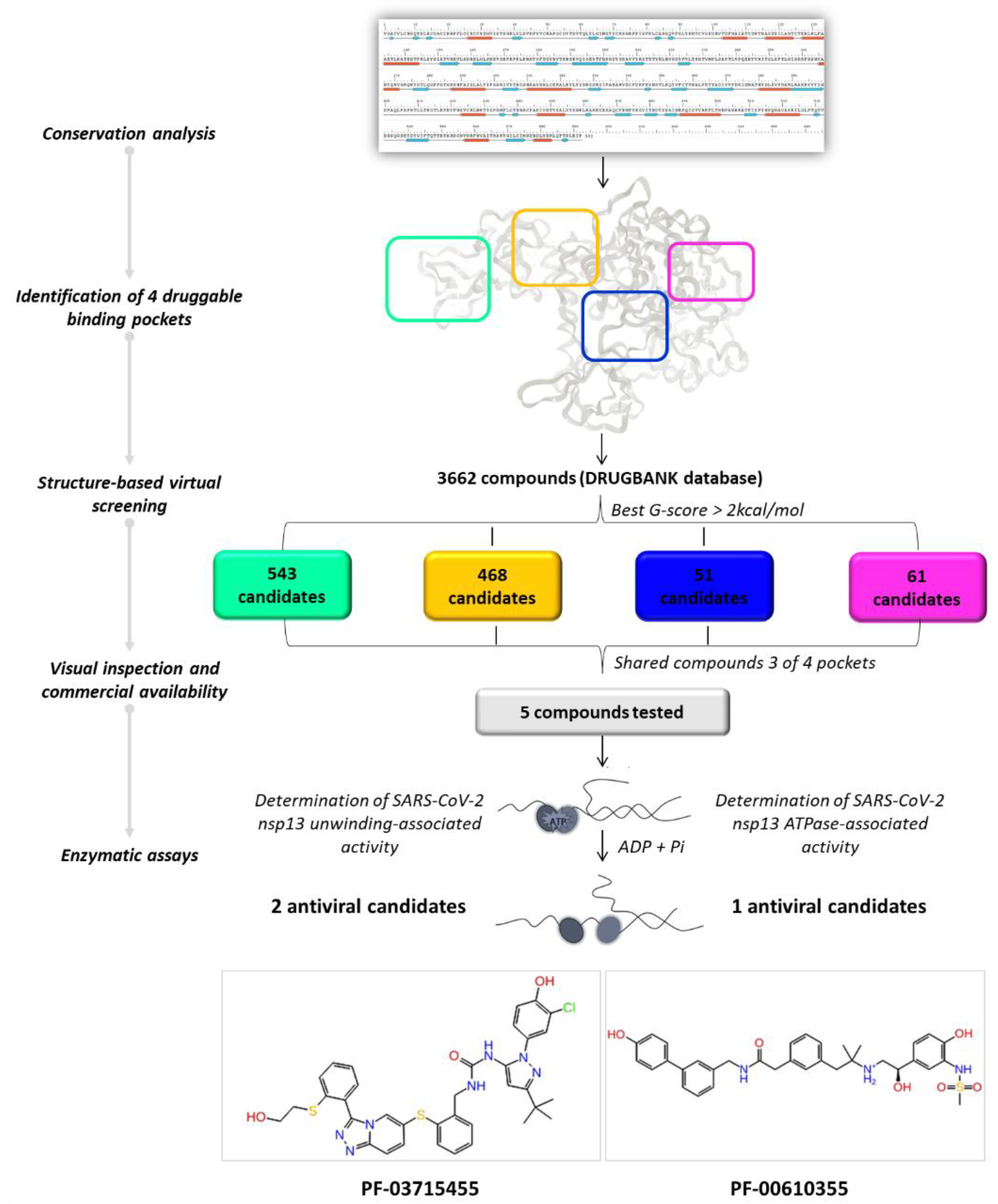
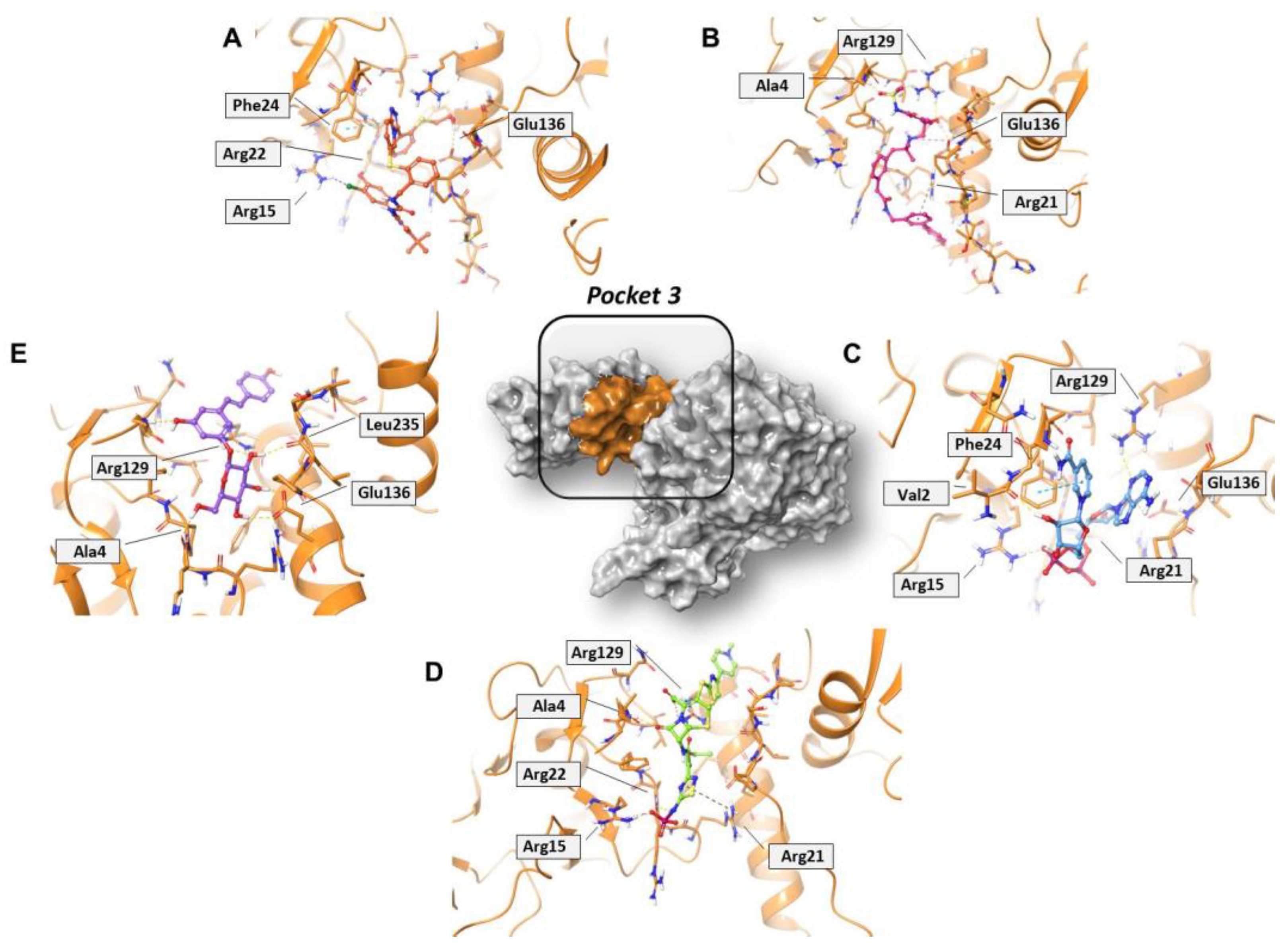
2.2. Identification of the Druggable Binding Pockets
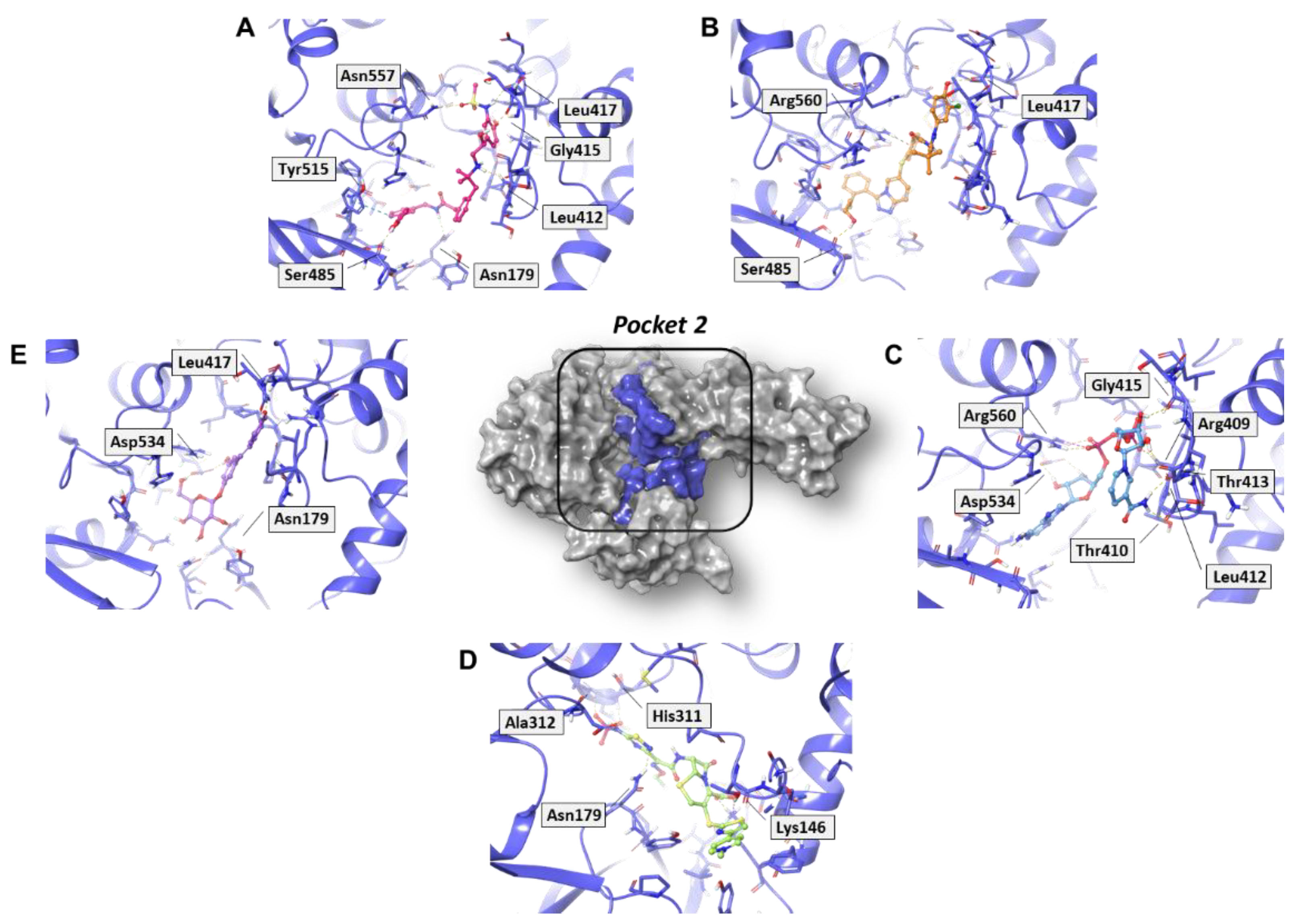
2.3. Residue Interaction Network Analysis of Helicase
2.4. In Silico Drug Repurposing
2.5. In Vitro Evaluation of Compounds Enzymatic Activity
3. Discussion
4. Materials and Methods
4.1. Conservation Analysis
4.2. Molecular Modeling
4.3. Structural Bioinformatics Analysis
4.4. SARS-CoV-2 nsp13 Expression and Purification
4.5. Determination of SARS-CoV-2 nsp13 Unwinding-Associated Activity
4.6. Determination of SARS-CoV-2 nsp13 ATPase-Associated Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- WHO COVID-19 Dashboard. World Health Organization: Geneva, Switzerland. 2020. Available online: https://covid19.who.int/ (accessed on 16 September 2022).
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Tillett, R.L.; Sevinsky, J.R.; Hartley, P.D.; Kerwin, H.; Crawford, N.; Gorzalski, A.; Laverdure, C.; Verma, S.C.; Rossetto, C.C.; Jackson, D.; et al. Genomic evidence for reinfection with SARS-CoV-2: A case study. Lancet Infect. Dis. 2021, 21, 52–58. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, T.; Islam, A.; Hassan, I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; AlAjmi, M.F.; Hussain, A.; Rehman, T.; Islam, A.; Hassan, I. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: Possible implication in COVID-19 therapy. Biosci Rep. 2020, 40, BSR20201256. [Google Scholar] [CrossRef]
- Imran, M.; Kumar Arora, M.; Asdaq, S.M.B.; Khan, S.A.; Alaqel, S.I.; Alshammari, M.K.; Alshehri, M.M.; Alshrari, A.S.; Mateq Ali, A.; Al-shammeri, A.M.; et al. Discovery, Development, and Patent Trends on Molnupiravir: A Prospective Oral Treatment for COVID-19. Molecules 2021, 26, 5795. [Google Scholar] [CrossRef]
- Khan, S.A.; Al-Balushi, K. Combating COVID-19: The role of drug repurposing and medicinal plants. J. Infect. Public Health 2021, 14, 495–503. [Google Scholar] [CrossRef]
- Imran, M.; Alshrari, A.S.; Asdaq, S.M.B.; Abida. Trends in the development of remdesivir based inventions against COVID-19 and other disorders: A patent review. J. Infect. Public Health 2021, 14, 1075–1086. [Google Scholar] [CrossRef]
- Ghasemnejad-Berenji, M.; Pashapour, S. Favipiravir and COVID-19: A Simplified Summary. Drug Res. 2021, 71, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Toots, M.; Yoon, J.J.; Cox, R.M.; Hart, M.; Sticher, Z.M.; Makhsous, N.; Plesker, R.; Barrena, A.H.; Reddy, P.G.; Mitchell, D.G.; et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019, 11, eaax5866. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Zhao, Y.; He, G.; Huang, W. A novel model of molnupiravir against SARS-CoV-2 replication: Accumulated RNA mutations to induce error catastrophe. Signal Transduct. Target Ther. 2021, 6, 410. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Alkhatib, M.; Salpini, R.; Carioti, L.; Ambrosio, F.A.; D’Anna, S.; Duca, L.; Costa, G.; Bellocchi, M.C.; Piermatteo, L.; Artese, A.; et al. Update on SARS-CoV-2 Omicron Variant of Concern and Its Peculiar Mutational Profile. Microbiol. Spectr. 2022, 10, e0273221. [Google Scholar] [CrossRef]
- Salpini, R.; Alkhatib, M.; Costa, G.; Piermatteo, L.; Ambrosio, F.A.; Di Maio, V.C.; Scutari, R.; Duca, L.; Berno, G.; Fabeni, L.; et al. Key genetic elements, single and in clusters, underlying geographically dependent SARS-CoV-2 genetic adaptation and their impact on binding affinity for drugs and immune control. J. Antimicrob. Chemother. 2021, 76, 396–412. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e13. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020, 11, 5874. [Google Scholar] [CrossRef]
- Ivanov, K.A.; Thiel, V.; Dobbe, J.C.; van der Meer, Y.; Snijder, E.J.; Ziebuhr, J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004, 78, 5619–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandão-Neto, J.; Dunnett, L.; Gorrie-Stone, T.; Skyner, R.; Fearon, D.; et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- Yue, K.; Yao, B.; Shi, Y.; Yang, Y.; Qian, Z.; Ci, Y.; Shi, L. The stalk domain of SARS-CoV-2 NSP13 is essential for its helicase activity. Biochem. Biophys. Res. Commun. 2022, 601, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Vishveshwara, S.; Brinda, K.; Kannan, N. Protein structure: Insights from graph theory. J. Theor. Comput. Chem. 2002, 1, 187–211. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Steed, M.E.; Rybak, M.J. Ceftaroline: A new cephalosporin with activity against resistant gram-positive pathogens. Pharmacotherapy 2010, 30, 375–389. [Google Scholar] [CrossRef]
- Lin, S.J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Du, Q.H.; Peng, C.; Zhang, H. Polydatin: A review of pharmacology and pharmacokinetics. Pharm. Biol. 2013, 51, 1347–1354. [Google Scholar] [CrossRef]
- Diderichsen, P.M.; Cox, E.; Martin, S.W.; Cleton, A.; Ribbing, J. Characterizing systemic exposure of inhaled drugs: Application to the long-acting β2-agonist PF-00610355. Clin. Pharmacokinet. 2013, 52, 443–452. [Google Scholar] [CrossRef]
- Norman, P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin. Investig. Drugs 2015, 24, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Adedeji, A.O.; Singh, K.; Calcaterra, N.E.; DeDiego, M.L.; Enjuanes, L.; Weiss, S.; Sarafianos, S.G. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 2012, 56, 4718–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, A.; Wycisk, K.; Talarico, C.; Manelfi, C.; Milia, J.; Cannalire, R.; Esposito, F.; Gribbon, P.; Zaliani, A.; Iaconis, D.; et al. Natural Compounds Inhibit SARS-CoV-2 nsp13 Unwinding and ATPase Enzyme Activities. ACS Pharmacol. Transl. Sci. 2022, 5, 226–239. [Google Scholar] [CrossRef]
- Ziebuhr, J. The coronavirus replicase. Curr. Top Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar]
- Yazdani, S.; De Maio, N.; Ding, Y.; Shahani, V.; Goldman, N.; Schapira, M. Genetic Variability of the SARS-CoV-2 Pocketome. J. Proteome Res. 2021, 20, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updates 2020, 53, 100721. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Abohashrh, M.; Baig, M.H.; Dong, J.J.; Alam, M.M.; Ahmad, I.; Irfan, S. Screening of drug databank against WT and mutant main protease of SARS-CoV-2: Towards finding potential compound for repurposing against COVID-19. Saudi J. Biol. Sci. 2021, 28, 3152–3159. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2018.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Available online: www.drugbank.ca. (accessed on 1 October 2020).
- LigPrep; Schrödinger, LLC: New York, NY, USA, 2018.
- MacroModel; Schrödinger, LLC: New York, NY, USA, 2018.
- Glide; Schrödinger, LLC: New York, NY, USA, 2018.
- Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2015.
- Kannan, N.; Vishveshwara, S. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 1999, 292, 441–464. [Google Scholar] [CrossRef]
- Brysbaert, G.; Lorgouilloux, K.; Vranken, W.F.; Lensink, M.F. RINspector: A Cytoscape app for centrality analyses and DynaMine flexibility prediction. Bioinformatics 2018, 34, 294–296. [Google Scholar] [CrossRef] [Green Version]
- Brysbaert, G.; Lensink, M.F. Centrality Measures in Residue Interaction Networks to Highlight Amino Acids in Protein-Protein Binding. Front. Bioinform. 2021, 1, 684970. [Google Scholar] [CrossRef]
- del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 2006.0019. [Google Scholar] [CrossRef] [Green Version]
- Looney, S.W.; Hagan, J.L. 4 statistical methods for assessing biomarkers and analyzing biomarker data. Handb. Stat. 2007, 27, 109–147. [Google Scholar]
- Abidi, S.H.; Almansour, N.M.; Amerzhanov, D.; Allemailem, K.S.; Rafaqat, W.; Ibrahim, M.A.A.; la Fleur, P.; Lukac, M.; Ali, S. Repurposing potential of posaconazole and grazoprevir as inhibitors of SARS-CoV-2 helicase. Sci. Rep. 2021, 11, 10290. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RCA | BCA | CCA | Z-RCA | |
|---|---|---|---|---|
| 10 | 5 (2.1·10−4) | 6 (2.5·10−4) | 5 (2.1·10−4) | 6 (2.5·10−4) |
| 20 | 10 (8.3·10−5) | 9 (5.1·10−4) | 10 (8.3·10−5) | 9 (5.1·10−4) |
| 50 | 19 (3.7·10−5) | 20 (8.9·10−6) | 19 (3.7·10−5) | 18 (1.4·10−4) |
| Chemical Group | Name | DrugBank ID | Structure | Pockets | |||
|---|---|---|---|---|---|---|---|
| 1 (−11.73 a) | 2 (−8.66 a) | 3 (−6.82 a) | 4 (−6.81 a) | ||||
| Acetamide derivatives | PF-00610355 | DB11871 | 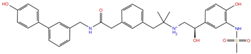 | −6.19 | −8.51 | −5.65 | −6.81 |
| Beta-lactam derivatives | Cefpiramide | DB00430 |  | −5.48 | −7.05 | −5.51 | −5.47 |
| Ceftaroline fosamil | DB06590 |  | −9.43 | −6.82 | −5.38 | −5.15 | |
| Glicoside derivatives | Acteoside | DB12996 |  | −6.05 | −8.13 | −4.83 | −5.49 |
| Polydatin | DB11263 | 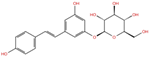 | −5.94 | −6.70 | −5.02 | −4.89 | |
| Rutin | DB01698 | 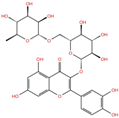 | −5.62 | −6.84 | −5.61 | −5.01 | |
| Phenol derivatives | Metaraminol | DB00610 | 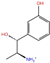 | −4.92 | −6.84 | −6.18 | −5.25 |
| Phosphono derivatives | Foscarnet | DB00171 | 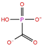 | −11.73 | −4.10 | −4.85 | −4.99 |
| Pteridine analogs | 5-methyltetrahydrofolic acid | DB04789 | 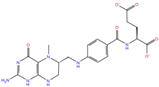 | −10.21 | −5.85 | −4.99 | −5.48 |
| Riboflavin | DB00140 |  | −5.80 | −6.68 | −4.86 | −5.44 | |
| Purine analogs | Inarigivir soproxil | DB15063 |  | −7.13 | −7.79 | −6.49 | −5.49 |
| NADH | DB00157 | 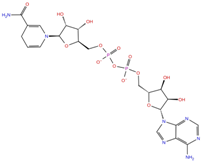 | −8.60 | −7.15 | −5.41 | −5.38 | |
| Regadenoson | DB06213 | 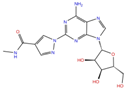 | −5.29 | −6.92 | −5.15 | −5.29 | |
| Triazole derivatives | PF-03715455 | DB12138 | 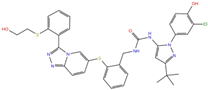 | −6.42 | −6.82 | −4.86 | −5.86 |
| Name | Developmental Phase | Pharmacological Data |
|---|---|---|
| Ceftaroline fosamil | Approved | Antibacterial activity [28] |
| NADH | Approved Nutraceutical | Nutritional and vitamin supplementation [29] |
| PF-03715455 | Investigational | In clinical trials for the treatment of asthma, pulmonary disease, chronic obstructive, and Chronic Obstructive Pulmonary Disease (COPD) ClinicalTrials.gov (NCT02219048) |
| PF-00610355 | Investigational | In clinical trials for the treatment of lung disease, pulmonary disease, asthma, and bronchial diseases ClinicalTrials.gov (NCT00783406) |
| Polydatin | Approved | Anti-inflammatory, immunoregulatory, anti-oxidative, and anti-tumor activities [30] |
| Compound | a IC50 (μM) Unwinding | b IC50 (μM) NTPase |
|---|---|---|
| Ceftaroline fosamil | >30 (100%) c | >30 (100%) |
| NADH | >30 (100%) | >30 (100%) |
| Polydatin | >30 (100%) | >30 (100%) |
| PF-03715455 | 3.02 ± 0.21 | 9.26 ± 1.93 |
| PF-00610355 | 22.4 ± 1.7 | >30 (100%) |
| SSYA10-001 | 2.3 ± 0.7 | ND d |
| Licoflavone C | 8.7 ± 1.3 | 20.9 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, I.; Ambrosio, F.A.; Costa, G.; Corona, A.; Alkhatib, M.; Salpini, R.; Lemme, S.; Vergni, D.; Svicher, V.; Santoro, M.M.; et al. Targeting SARS-CoV-2 nsp13 Helicase and Assessment of Druggability Pockets: Identification of Two Potent Inhibitors by a Multi-Site In Silico Drug Repurposing Approach. Molecules 2022, 27, 7522. https://doi.org/10.3390/molecules27217522
Romeo I, Ambrosio FA, Costa G, Corona A, Alkhatib M, Salpini R, Lemme S, Vergni D, Svicher V, Santoro MM, et al. Targeting SARS-CoV-2 nsp13 Helicase and Assessment of Druggability Pockets: Identification of Two Potent Inhibitors by a Multi-Site In Silico Drug Repurposing Approach. Molecules. 2022; 27(21):7522. https://doi.org/10.3390/molecules27217522
Chicago/Turabian StyleRomeo, Isabella, Francesca Alessandra Ambrosio, Giosuè Costa, Angela Corona, Mohammad Alkhatib, Romina Salpini, Saverio Lemme, Davide Vergni, Valentina Svicher, Maria Mercedes Santoro, and et al. 2022. "Targeting SARS-CoV-2 nsp13 Helicase and Assessment of Druggability Pockets: Identification of Two Potent Inhibitors by a Multi-Site In Silico Drug Repurposing Approach" Molecules 27, no. 21: 7522. https://doi.org/10.3390/molecules27217522