
Identification of Boronate-Containing Diarylpyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors


 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Chemistry
2.2. Anti-HIV-1 Activity Evaluation
2.3. HIV-1 RT Inhibition Assay
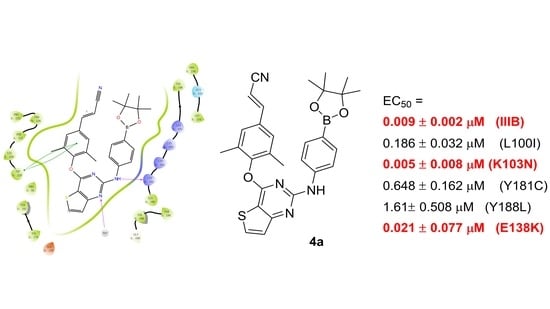
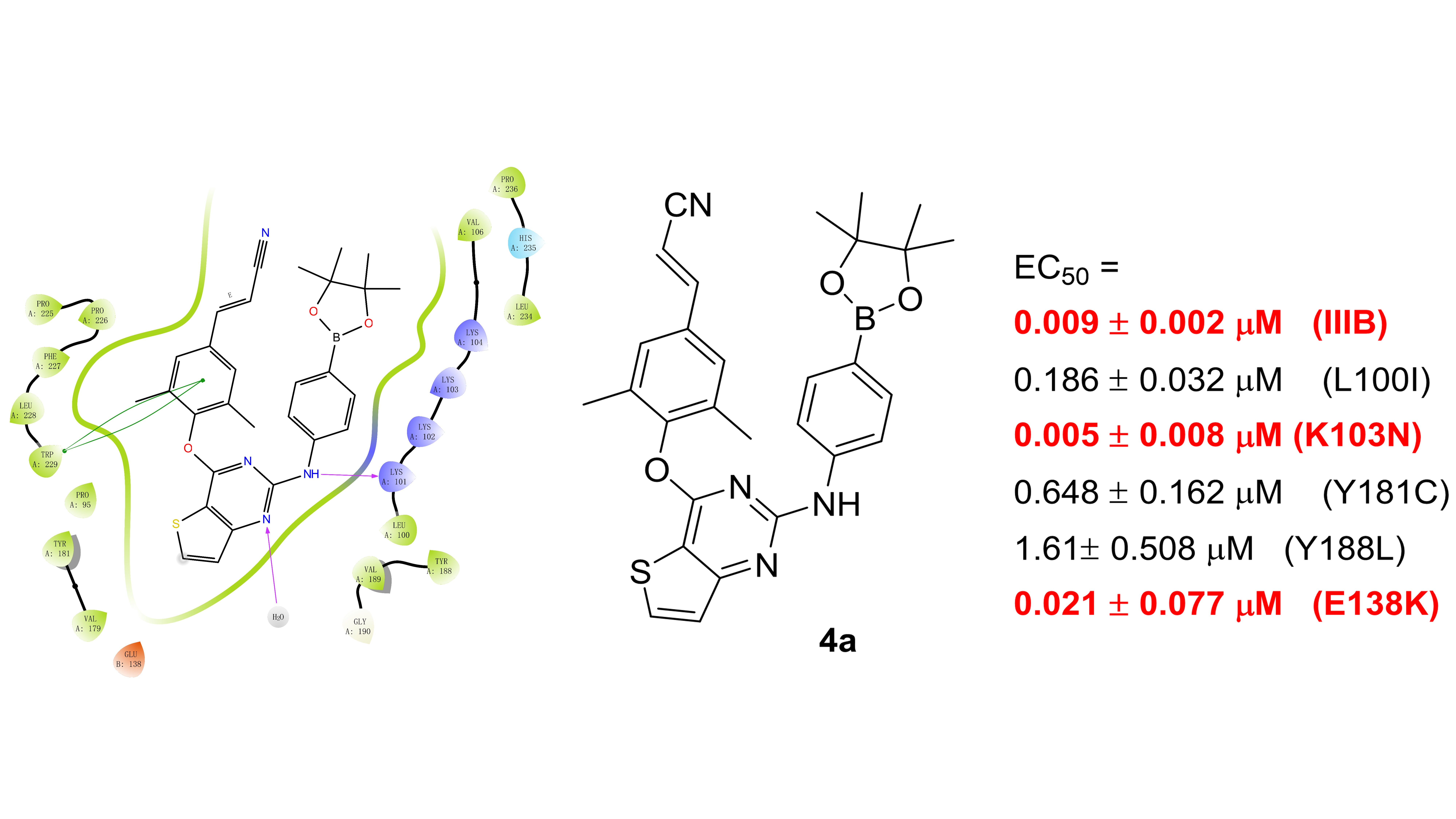
2.4. Molecular Docking (MD) Simulation
2.5. In Silico Prediction of Physicochemical Properties
3. Materials and Methods
3.1. Synthesis
3.1.1. General Procedure for the Preparation of Target Compounds 3a–j and 4a–c
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) thieno [3,2-d]pyrimidin-4-yl)oxy)benzonitrile (3a) (72%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) thieno [2,3-d]pyrimidin-4-yl)oxy)benzonitrile (3b) (65%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) -5,7-dihydrofuro [3,4-d]pyrimidin-4-yl)oxy)benzonitrile (3c) (62%, yield)
3,5-Dimethyl-4-((5-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) thiazolo [4,5-d]pyrimidin-7-yl)oxy)benzonitrile (3d) (72%, yield)
3,5-Dimethyl-4-((5-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) thiazolo [5,4-d]pyrimidin-7-yl)oxy)benzonitrile (3e) (66%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) pyrimidin-4-yl)oxy)benzonitrile (3f) (75%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) quinazolin-4-yl)oxy)benzonitrile (3g) (60%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) pyrido [3,2-d]pyrimidin-4-yl)oxy)benzonitrile (3h) (52%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) -5,6,7,8-tetrahydroquinazolin-4-yl)oxy)benzonitrile (3i) (63%, yield)
3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)amino) -7,8-dihydro-6H-thiopyrano [3,2-d]pyrimidin-4-yl)oxy)benzonitrile (3j) (59%, yield)
(E)-3-(3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl) amino)thieno [3,2-d]pyrimidin-4-yl)oxy)phenyl)acrylonitrile (4a) (72%, yield)
(E)-3-(3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl) amino)thieno [2,3-d]pyrimidin-4-yl)oxy)phenyl)acrylonitrile (4b) (70%, yield)
(E)-3-(3,5-Dimethyl-4-((2-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl) amino)-5,7-dihydrofuro [3,4-d]pyrimidin-4-yl)oxy)phenyl)acrylonitrile (4c) (66%, yield)
3.2. In Vitro Anti-HIV Activities Assays in MT-4 Cells
3.3. HIV-1 RT Inhibition Assays
3.4. Molecular Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fauci, A.S.; Lane, H.C. Four decades of HIV/AIDS—Much accomplished, much to do. N. Engl. J. Med. 2020, 383, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Battini, L.; Bollini, M. Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2019, 39, 1235–1273. [Google Scholar] [CrossRef]
- Feng, D.; Zuo, X.; Jing, L.; Chen, C.H.; Olotu, F.A.; Lin, H.; Soliman, M.; De Clercq, E.; Pannecouque, C.; Lee, K.H.; et al. Design, synthesis, and evaluation of “dual-site”-binding diarylpyrimidines targeting both NNIBP and the NNRTI adjacent site of the HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2021, 211, 113063. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV drug discovery and development: Current innovations and future trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef]
- Namasivayam, V.; Vanangamudi, M.; Kramer, V.G.; Kurup, S.; Zhan, P.; Liu, X.; Kongsted, J.; Byrareddy, S.N. The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J. Med. Chem. 2019, 62, 4851–4883. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Elsulfavirine: First global approval. Drugs 2017, 77, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.S.; Luo, R.H.; Hu, X.L.; Chen, H.; Xiang, S.Y.; Tang, C.R.; Zhang, C.T.; Shen, X.N.; Zheng, Y.T. The new NNRTI ACC007 combined with lamivudine and tenofovir disoproxil fumarate show synergy anti-HIV activity in vitro. Curr. HIV Res. 2020, 18, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding drug resistance in HIV reverse transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Lehman, D.A.; Wamalwa, D.C.; McCoy, C.O.; Matsen, F.A.; Langat, A.; Chohan, B.H.; Benki-Nugent, S.; Custers-Allen, R.; Bushman, F.D.; John-Stewart, G.C.; et al. Low-frequency nevirapine resistance at multiple sites may predict treatment failure in infants on nevirapine-based treatment. J. Acquir. Immune Defic. Syndr. 2012, 60, 225–233. [Google Scholar] [CrossRef]
- Beyrer, C.; Pozniak, A. HIV drug resistance - an emerging threat to epidemic control. N. Engl. J. Med. 2017, 377, 1605–1607. [Google Scholar] [CrossRef]
- Kang, D.; Ruiz, F.X.; Sun, Y.; Feng, D.; Jing, L.; Wang, Z.; Zhang, T.; Gao, S.; Sun, L.; De Clercq, E.; et al. 2,4,5-Trisubstituted pyrimidines as potent HIV-1 NNRTIs: Rational design, synthesis, activity evaluation, and crystallographic studies. J. Med. Chem. 2021, 64, 4239–4256. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef] [PubMed]
- Plescia, J.; Moitessier, N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 2020, 195, 112270. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef]
- Windsor, I.W.; Palte, M.J.; Lukesh, J.C., 3rd; Gold, B.; Forest, K.T.; Raines, R.T. Sub-picomolar inhibition of HIV-1 protease with a boronic acid. J. Am. Chem. Soc. 2018, 140, 14015–14018. [Google Scholar] [CrossRef]
- Chong, P.Y.; Shotwell, J.B.; Miller, J.; Price, D.J.; Maynard, A.; Voitenleitner, C.; Mathis, A.; Williams, S.; Pouliot, J.J.; Creech, K.; et al. Design of N-benzoxaborole benzofuran GSK8175-optimization of human pharmacokinetics inspired by metabolites of a failed clinical HCV inhibitor. J. Med. Chem. 2019, 62, 3254–3267. [Google Scholar] [CrossRef]
- Zhan, P.; Kang, D.; Liu, X. Resurrecting the condemned: Identification of n-benzoxaborole benzofuran GSK8175 as a clinical candidate with reduced metabolic liability. J. Med. Chem. 2019, 62, 3251–3253. [Google Scholar] [CrossRef]
- Feng, D.; Wei, F.; Sun, Y.; Sharma, P.P.; Zhang, T.; Lin, H.; Rathi, B.; De Clercq, E.; Pannecouque, C.; Kang, D.; et al. Boronic acid-containing diarylpyrimidine derivatives as novel HIV-1 NNRTIs: Design, synthesis and biological evaluation. Chin. Chem. Lett. 2021, 32, 4053–4057. [Google Scholar] [CrossRef]
- Xu, S.; Song, S.; Sun, L.; Gao, P.; Gao, S.; Ma, Y.; Kang, D.; Cheng, Y.; Zhang, X.; Cherukupalli, S.; et al. Indolylarylsulfones bearing phenylboronic acid and phenylboronate ester functionalities as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2022, 53, 116531. [Google Scholar]
- Kang, D.; Fang, Z.; Li, Z.; Huang, B.; Zhang, H.; Lu, X.; Xu, H.; Zhou, Z.; Ding, X.; Daelemans, D.; et al. Design, synthesis, and evaluation of thiophene[3,2-d]pyrimidine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors with significantly improved drug resistance profiles. J. Med. Chem. 2016, 59, 7991–8007. [Google Scholar]
- Kang, D.; Fang, Z.; Huang, B.; Lu, X.; Zhang, H.; Xu, H.; Huo, Z.; Zhou, Z.; Yu, Z.; Meng, Q.; et al. Structure-based optimization of thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with improved potency against resistance-associated variants. J. Med. Chem. 2017, 60, 4424–4443. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cherukupalli, S.; Xie, M.; Wang, W.; Jiang, X.; Jia, R.; Pannecouque, C.; De Clercq, E.; Kang, D.; Zhan, P.; et al. Contemporary Medicinal Chemistry Strategies for the Discovery and Development of Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem. 2022, 65, 3729–3757. [Google Scholar] [CrossRef]
- Feng, D.; Wei, F.; Wang, Z.; Kang, D.; Zhan, P.; Liu, X. Development of a practical synthesis of etravirine via a microwave-promoted amination. Chem. Cent. J. 2018, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Feng, D.; Ginex, T.; Zou, J.; Wei, F.; Zhao, T.; Huang, B.; Sun, Y.; Desta, S.; De Clercq, E.; et al. Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm. Sin. B 2020, 10, 878–894. [Google Scholar] [CrossRef]
- Kang, D.; Feng, D.; Sun, Y.; Fang, Z.; Wei, F.; De Clercq, E.; Pannecouque, C.; Liu, X.; Zhan, P. Structure-based bioisosterism yields HIV-1 NNRTIs with improved drug-resistance profiles and favorable pharmacokinetic properties. J. Med. Chem. 2020, 63, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyter, J.; De Clercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 1988, 20, 309–321. [Google Scholar] [CrossRef]
- Pannecouque, C.; Daelemans, D.; De Clercq, E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: Revisited 20 years later. Nat. Protoc. 2008, 3, 427–434. [Google Scholar] [CrossRef]
- Suzuki, K.; Craddock, B.P.; Okamoto, N.; Kano, T.; Steigbigel, R.T. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J. Virol. Methods. 1993, 44, 189–198. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zhang, H.; Wang, Z.; Zhao, T.; Ginex, T.; Luque, F.J.; Yang, Y.; Wu, G.; Feng, D.; Wei, F.; et al. Identification of Dihydrofuro[3,4-d]pyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Promising Antiviral Activities and Desirable Physicochemical Properties. J. Med. Chem. 2019, 62, 1484–1501. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Sun, Y.; Feng, D.; Gao, S.; Wang, Z.; Jing, L.; Zhang, T.; Jiang, X.; Lin, H.; De Clercq, E.; et al. Development of Novel Dihydrofuro[3,4-d]pyrimidine Derivatives as HIV-1 NNRTIs to Overcome the Highly Resistant Mutant Strains F227L/V106A and K103N/Y181C. J. Med. Chem. 2022, 65, 2458–2470. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compds. | R | Center Core | EC50 (μM) a | CC50 (μM) b | SI c (RF) d | ||
|---|---|---|---|---|---|---|---|
| IIIB | RES056 | IIIB | RES056 | ||||
| 3a | CN |  | 0.064 ± 0.021 | >27.4 | 27.4 ± 1.20 | 431 (1.0) | <1 (>431) |
| 3b | CN |  | 1.30 ± 0.275 | 174 ± 34.5 | >254 | >193 (1.0) | >1 (134) |
| 3c | CN |  | 0.051 ± 0.015 | 25.8 ± 7.93 | 94.8 ± 32.2 | 1847 (1.0) | <1 (503) |
| 3d | CN |  | 0.120 ± 0.056 | >24.1 | 24.1 ± 3.76 | 201 (1.0) | <1 (>201) |
| 3e | CN |  | 0.215 ± 0.091 | >26.5 | 26.5 ± 1.78 | 123 (1.0) | <1 (>123) |
| 3f | CN |  | 0.149 ± 0.049 | >30.6 | 30.6 ± 1.92 | 206 (1.0) | <1 (>206) |
| 3g | CN |  | 6.17 ± 1.83 | >254 | >254 | >41 (1.0) | X1 e (>41.1) |
| 3h | CN |  | 0.112 ± 0.024 | >26.0 | 26.0 ± 2.33 | 232 (1.0) | <1 (>232) |
| 3i | CN |  | 0.191 ± 0.088 | >27.7 | 27.7 ± 1.27 | 145 (1.0) | <1 (>145) |
| 3j | CN |  | 0.141 ± 0.053 | >25.9 | 25.9 ± 1.41 | 184 (1.0) | <1 (>184) |
| 4a | CV |  | 0.009 ± 0.002 | >238 | >238 | >27,516 (1.0) | X1 e (>27516) |
| 4b | CV |  | 0.010 ± 0.003 | 2.30 ± 0.660 | 22.1 ± 3.69 | 2161 (1.0) | 10 (224) |
| 4c | CV |  | 0.014 ± 0.006 | 13.9 ± 9.32 | 80.3 ± 31.3 | 5890 (1.0) | 6 (1018) |
| NVP | - | - | 0.151 ± 0.037 | >15.0 | >15.0 | >100 (1.0) | X1 e (99.9) |
| EFV | - | - | 0.003 ± 0.001 | 0.337 ± 0.149 | >6.35 | >2268 (1.0) | >19 (120) |
| ETR | - | - | 0.004 ± 0.001 | 0.061 ± 0.034 | >4.60 | >1105 (1.0) | >76 (14.6) |
| Compds. | EC50 (μM) a | |||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | |
| 3a | 2.51 ± 0.155 | 0.081 ± 0.008 | 6.29 ± 3.27 | 6.02 ± 3.14 | 0.193 ± 0.077 | 4.95 ± 1.98 |
| 3c | 2.43 ± 0.302 | 0.120 ± 0.019 | 4.37 ± 2.19 | 4.52 ± 1.54 | 0.130 ± 0.041 | 9.10 ± 2.64 |
| 4a | 0.186 ± 0.032 | 0.005 ± 0.006 | 0.648 ± 0.162 | 1.61 ± 0.508 | 0.021 ± 0.007 | >1.81 |
| 4b | 0.280 ± 0.055 | 0.008 ± 0.002 | 0.675 ± 0.188 | 2.52 ± 0.238 | 0.030 ± 0.009 | 3.21 ± 1.97 |
| 4c | 0.553 ± 0.152 | 0.026 ± 0.005 | 1.28 ± 0.489 | 3.96 ± 0.894 | 0.039 ± 0.018 | 5.88 ± 2.52 |
| NVP | 0.623 ± 0.277 | 3.93 ± 1.01 | 5.06 ± 0.922 | 9.08 ± 2.41 | 0.168 ± 1.077 | 8.05 ± 3.54 |
| EFV | 0.032 ± 0.008 | 0.071 ± 0.020 | 0.005 ± 0.002 | 0.261 ± 0.118 | 0.006 ± 0.002 | 0.381 ± 0.249 |
| ETR | 0.008 ± 0.002 | 0.003 ± 0.008 | 0.016 ± 0.005 | 0.019 ± 0.007 | 0.010 ± 0.005 | 0.026 ± 0.017 |
| Compds. | SI a (RF) b | |||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | |
| 3a | 11 (39.4) | 341 (1.3) | 4 (98.8) | 5 (94.7) | 142 (3.0) | 6 (77.8) |
| 3c | 39 (47.3) | 792 (2.3) | 22 (85.1) | 21 (87.9) | 731 (2.5) | 10 (177.2) |
| 4a | >1285 (21.4) | >47,170 (0.6) | >368 (74.8) | >148 (506.8) | >11,261 (2.4) | X1 c (209.7) |
| 4b | 79 (27.4) | 2866 (0.8) | 33 (65.9) | 9 (246.1) | 732 (3.0) | 7 (313.5) |
| 4c | 145 (40.6) | 3110 (1.9) | 63 (94.2) | 20 (290.3) | 2036 (2.9) | 14 (431.5) |
| NVP | >24 (4.1) | >4 (26.1) | >3 (33.6) | >2 (60.4) | >89 (1.1) | >2 (53.5) |
| EFV | >200 (11.3) | >89 (25.4) | >1260 (1.8) | >24 (93.1) | >1149 (2.0) | >17 (136.2) |
| ETR | >549 (2.0) | >1417 (0.8) | >297 (3.7) | >248 (4.5) | >469 (2.4) | >178 (6.2) |
| Compds. | IC50 (μM) a | Compds. | IC50 (μM) a |
|---|---|---|---|
| 3a | 0.354 ± 0.043 | 4c | 0.200 ± 0.028 |
| 3c | 0.228 ± 0.045 | NVP | 0.638 ± 0.269 |
| 4a | 0.167 ± 0.031 | EFV | 0.008 ± 0.003 |
| 4b | 0.047 ± 0.001 | ETR b | 0.011 ± 0.000 |
| Compds. | nHA | nHD | TPSA | nRot | LogP |
|---|---|---|---|---|---|
| 4a | 7 | 1 | 92.52 | 5 | 6.549 |
| 4b | 7 | 1 | 92.52 | 5 | 6.707 |
| ETR | 7 | 3 | 124.6 | 3 | 3.47 |
| RPV | 6 | 2 | 103.88 | 3 | 3.47 |
| Optimal range | 0–12 | 0–7 | 0–140 | 0–11 | 0–3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, D.; Lin, H.; Jiang, L.; Wang, Z.; Sun, Y.; Zhou, Z.; Clercq, E.D.; Pannecouque, C.; Kang, D.; Zhan, P.; et al. Identification of Boronate-Containing Diarylpyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules 2022, 27, 7538. https://doi.org/10.3390/molecules27217538
Feng D, Lin H, Jiang L, Wang Z, Sun Y, Zhou Z, Clercq ED, Pannecouque C, Kang D, Zhan P, et al. Identification of Boronate-Containing Diarylpyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules. 2022; 27(21):7538. https://doi.org/10.3390/molecules27217538
Chicago/Turabian StyleFeng, Da, Hao Lin, Liyang Jiang, Zhao Wang, Yanying Sun, Zhongxia Zhou, Erik De Clercq, Christophe Pannecouque, Dongwei Kang, Peng Zhan, and et al. 2022. "Identification of Boronate-Containing Diarylpyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors" Molecules 27, no. 21: 7538. https://doi.org/10.3390/molecules27217538
APA StyleFeng, D., Lin, H., Jiang, L., Wang, Z., Sun, Y., Zhou, Z., Clercq, E. D., Pannecouque, C., Kang, D., Zhan, P., & Liu, X. (2022). Identification of Boronate-Containing Diarylpyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules, 27(21), 7538. https://doi.org/10.3390/molecules27217538