
Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro
 , , , , and
, , , , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
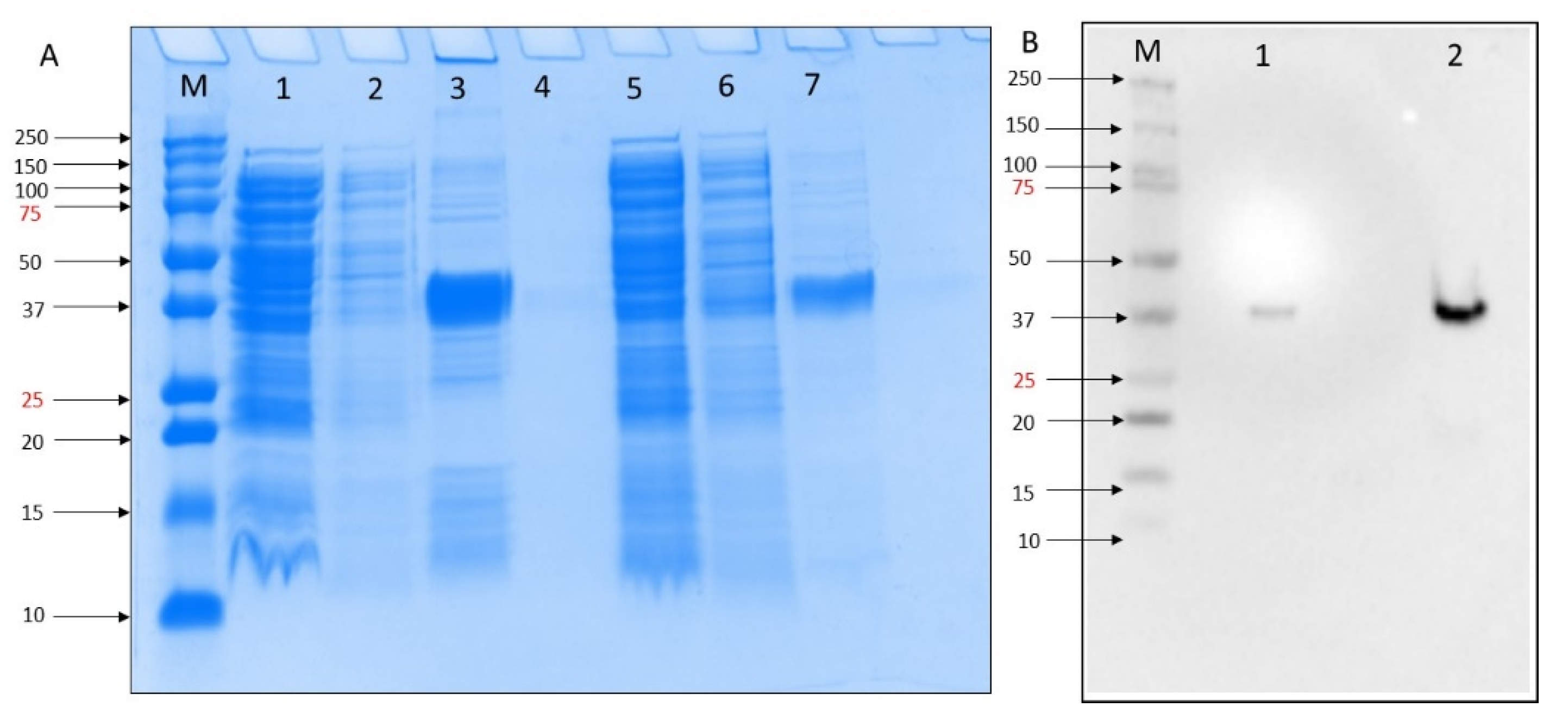
2.1. Expression and Purification of MERS-CoV PLpro
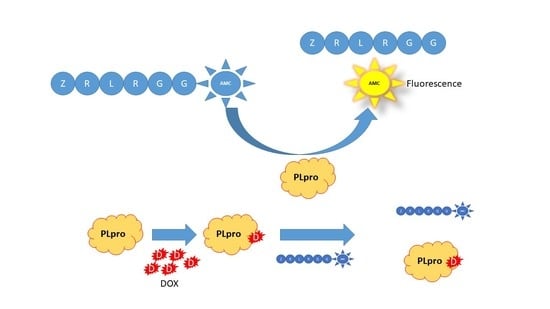
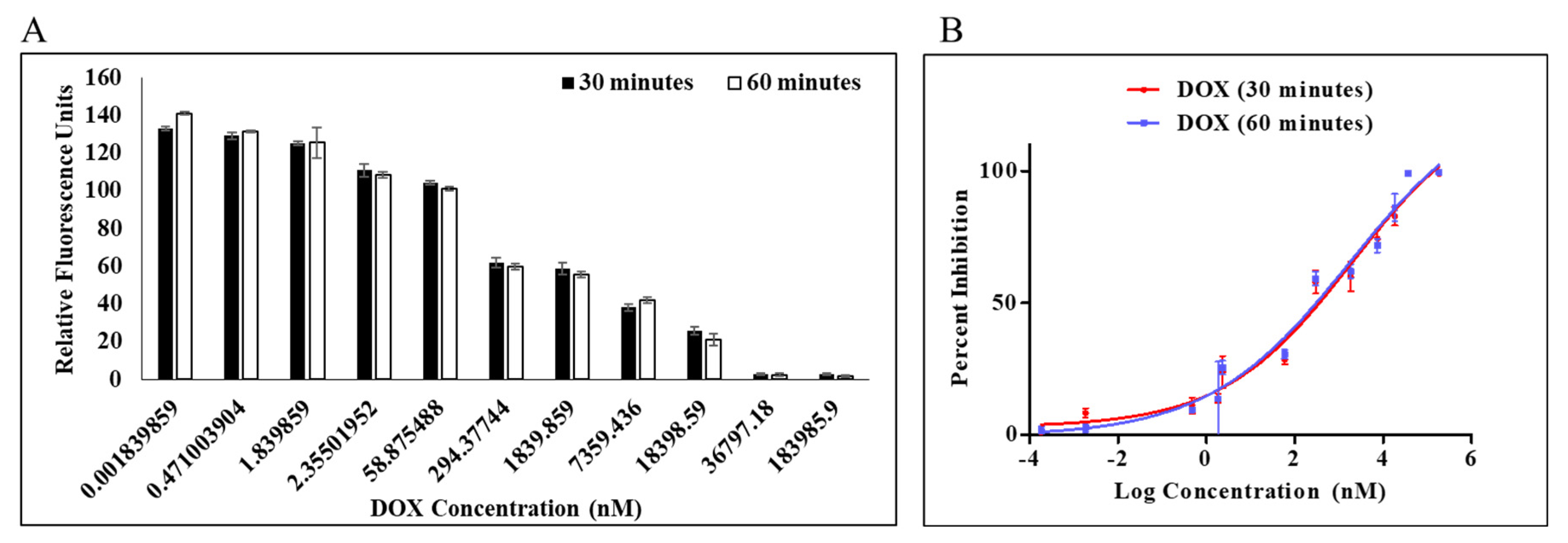
2.2. MERS-CoV PLpro Inhibition Assay
2.3. Thermal Shift Assay (TSA)
2.4. Doxorubicin Binding Site
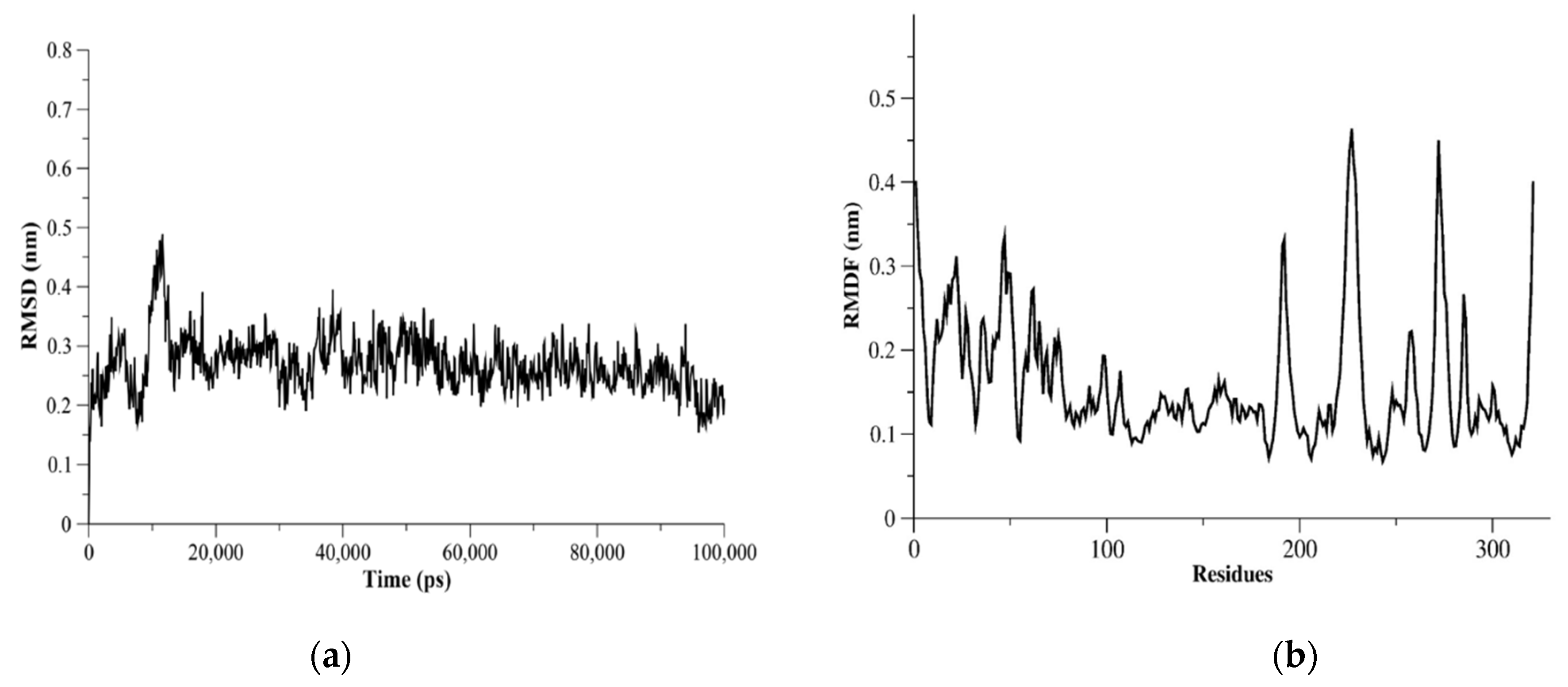
2.5. MERS-CoV PLpro Flexibility
3. Material and Method
3.1. Chemicals
3.2. Protease Expression Plasmids
3.3. SDS-PAGE
3.4. Western Blotting
3.5. Protein Purification
3.6. MERS-CoV PLpro Inhibition Assay
3.7. Thermal Shift Assay (TSA)
3.8. Molecular Dynamic (MD) Simulations
3.9. Docking of Doxorubicin to MERS-CoV PLpro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus Envelope Protein: Current Knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef] [Green Version]
- WHO. Middle East Respiratory Syndrome Coronavirus (MERS-CoV). 2022. Available online: https://www.who.int/health-topics/middle-east-respiratory-syndrome-coronavirus-mers#tab=tab_1 (accessed on 2 April 2022).
- McIntosh, K.; Hirsch, M.S.; Allyson Bloom, M. Coronavirus Disease 2019 (COVID-19). UpToDate 2020, 2019, 1–27. [Google Scholar]
- Wernery, U.; Rasoul, I.H.E.; Wong, E.Y.M.; Joseph, M.; Chen, Y.; Jose, S.; Tsang, A.K.L.; Patteril, N.A.G.; Chen, H.; Elizabeth, S.K.; et al. A Phylogenetically Distinct Middle East Respiratory Syndrome Coronavirus Detected in a Dromedary Calf from a Closed Dairy Herd in Dubai with Rising Seroprevalence with Age. Emerg. Microbes Infect. 2015, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? Int. J. Mol. Sci. 2017, 18, 2138. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lei, H.; Santarsiero, B.D.; Gatuz, J.L.; Cao, S.; Rice, A.J.; Patel, K.; Szypulinski, M.Z.; Ojeda, I.; Ghosh, A.K.; et al. Inhibitor Recognition Specificity of MERS-CoV Papain-like Protease May Differ from That of SARS-CoV. ACS Chem. Biol. 2015, 10, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.W.; Cheng, S.C.; Lai, H.Y.; Chou, C.Y. Differential Domain Structure Stability of the Severe Acute Respiratory Syndrome Coronavirus Papain-like Protease. Arch. Biochem. Biophys. 2012, 520, 74–80. [Google Scholar] [CrossRef]
- Lei, J.; Mesters, J.R.; Drosten, C.; Anemüller, S.; Ma, Q.; Hilgenfeld, R. Crystal Structure of the Papain-like Protease of MERS Coronavirus Reveals Unusual, Potentially Druggable Active-Site Features. Antivir. Res. 2014, 109, 72–82. [Google Scholar] [CrossRef]
- Johnson, V.A.; Calvez, V.; Günthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Richman, D.D. 2011 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar]
- Hulskotte, E.; Gupta, S.; Xuan, F.; van Zutven, M.; O’Mara, E.; Feng, H.P.; Wagner, J.; Butterton, J. Pharmacokinetic Interaction between the Hepatitis C Virus Protease Inhibitor Boceprevir and Cyclosporine and Tacrolimus in Healthy Volunteers. Hepatology 2012, 56, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Kwo, P.Y.; Lawitz, E.J.; McCone, J.; Schiff, E.R.; Vierling, J.M.; Pound, D.; Davis, M.N.; Galati, J.S.; Gordon, S.C.; Ravendhran, N.; et al. Efficacy of Boceprevir, an NS3 Protease Inhibitor, in Combination with Peginterferon Alfa-2b and Ribavirin in Treatment-Naive Patients with Genotype 1 Hepatitis C Infection (SPRINT-1): An Open-Label, Randomised, Multicentre Phase 2 Trial. Lancet 2010, 376, 705–716. [Google Scholar] [CrossRef]
- Steuer, C.; Heinonen, K.H.; Kattner, L.; Klein, C.D. Optimization of Assay Conditions for Dengue Virus Protease: Effect of Various Polyols and Nonionic Detergents. SLAS Discov. 2009, 14, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C.; Steuer, C.; Klein, C.D. Arylcyanoacrylamides as Inhibitors of the Dengue and West Nile Virus Proteases. Bioorganic Med. Chem. 2011, 19, 7318–7337. [Google Scholar] [CrossRef]
- Kandeel, M.; Abdelrahman, A.H.M.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A.A. Repurposing of FDA-Approved Antivirals, Antibiotics, Anthelmintics, Antioxidants, and Cell Protectives against SARS-CoV-2 Papain-like Protease. J. Biomol. Struct. Dyn. 2021, 39, 5129–5136. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV Papain-like Protease Has DeISGylating and Deubiquitinating Activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The Complex Structure of GRL0617 and SARS-CoV-2 PLpro Reveals a Hot Spot for Antiviral Drug Discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Lim, C.T.; Tan, K.W.; Wu, M.; Ulferts, R.; Armstrong, L.A.; Ozono, E.; Drury, L.S.; Milligan, J.C.; Zeisner, T.U.; Zeng, J.; et al. Identifying SARS-CoV-2 Antiviral Compounds by Screening for Small Molecule Inhibitors of Nsp3 Papain-like Protease. Biochem. J. 2021, 478, 2517–2531. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, K.; Pan, J.; Lei, Y.; Zhang, D.; Fang, L.; Tang, J.; Chen, X.; Ma, Y.; Zheng, Y.; et al. Repurposing Clinically Approved Drugs for COVID-19 Treatment Targeting SARS-CoV-2 Papain-like Protease. Int. J. Biol. Macromol. 2021, 188, 137–146. [Google Scholar] [CrossRef]
- Zhao, Y.; Du, X.; Duan, Y.; Pan, X.; Sun, Y.; You, T.; Han, L.; Jin, Z.; Shang, W.; Yu, J.; et al. High-Throughput Screening Identifies Established Drugs as SARS-CoV-2 PLpro Inhibitors. Protein Cell 2021, 12, 877–888. [Google Scholar] [CrossRef]
- Bafna, K.; White, K.; Harish, B.; Rosales, R.; Ramelot, T.A.; Acton, T.B.; Moreno, E.; Kehrer, T.; Miorin, L.; Royer, C.A.; et al. Hepatitis C Virus Drugs That Inhibit SARS-CoV-2 Papain-like Protease Synergize with Remdesivir to Suppress Viral Replication in Cell Culture. Cell Rep. 2021, 35, 109133. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.E.; Conigliaro, J.; Wang, T.C.; Tracey, K.J.; Callahan, M.V.; Abrams, J.A. Famotidine Use Is Associated With Improved Clinical Outcomes in Hospitalized COVID-19 Patients: A Propensity Score Matched Retrospective Cohort Study. Gastroenterology 2020, 159, 1129–1131.e3. [Google Scholar] [CrossRef] [PubMed]
- Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors|Nature. Available online: https://www.nature.com/articles/s41586-020-2223-y (accessed on 25 October 2022).
- PubChem Doxorubicin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/31703 (accessed on 16 September 2022).
- Fornari, F.A.; Randolph, J.K.; Yalowich, J.C.; Ritke, M.K.; Gewirtz, D.A. Interference by Doxorubicin with DNA Unwinding in MCF-7 Breast Tumor Cells. Mol. Pharmacol. 1994, 45, 649. [Google Scholar] [PubMed]
- Momparler, R.L.; Karon, M.; Siegel, S.E.; Avila, F. Effect of Adriamycin on DNA, RNA, and Protein Synthesis in Cell-Free Systems and Intact Cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar] [PubMed]
- Sritharan, S.; Sivalingam, N. A Comprehensive Review on Time-Tested Anticancer Drug Doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Al-Motawa, M.S.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 Virus to Proteotoxicity—Opportunity for Repurposed Chemotherapy of COVID-19 Infection. Front. Pharmacol. 2020, 11, 585408. [Google Scholar] [CrossRef] [PubMed]
- Sajid Jamal, Q.M.; Alharbi, A.H.; Ahmad, V. Identification of Doxorubicin as a Potential Therapeutic against SARS-CoV-2 (COVID-19) Protease: A Molecular Docking and Dynamics Simulation Studies. J. Biomol. Struct. Dyn. 2021. [Google Scholar] [CrossRef]
- Chiu, W.; Verschueren, L.; Eynde, C.; Buyck, C.; Meyer, S.; Jochmans, D.; Bojkova, D.; Ciesek, S.; Cinatl, J.; Jonghe, S.; et al. Development and Optimization of a High-Throughput Screening Assay for in Vitro Anti-SARS-CoV-2 Activity: Evaluation of 5676 Phase 1 Passed Structures. J. Med. Virol. 2022, 94, 3101–3111. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [Green Version]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions That Promote Protein Stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Ericsson, U.B.; Hallberg, B.M.; DeTitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-Based High-Throughput Stability Optimization of Proteins for Structural Studies. Anal. Biochem. 2006, 357, 289–298. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. SLAS Discov. 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Cimmperman, P.; Baranauskiene, L.; Jachimovičiute, S.; Jachno, J.; Torresan, J.; Michailoviene, V.; Matuliene, J.; Sereikaite, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef]
- Debeljak, N.; Feldman, L.; Davis, K.L.; Komel, R.; Sytkowski, A.J. Variability in the Immunodetection of His-Tagged Recombinant Proteins. Anal. Biochem. 2006, 359, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Shahid, M.; Raish, M.; Ahmad, A.; Bin Jardan, Y.A.; Ansari, M.A.; Ahad, A.; Alkharfy, K.M.; Alaofi, A.L.; Al-jenoobi, F.I. Sinapic Acid Ameliorates Acetic Acid-Induced Ulcerative Colitis in Rats by Suppressing Inflammation, Oxidative Stress, and Apoptosis. Molecules 2022, 27, 4139. [Google Scholar]
- Huynh, K.; Partch, C.L. Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. Curr. Protoc. Protein Sci. 2015, 79, 28.9.1–28.9.14. [Google Scholar] [CrossRef] [Green Version]
- Alaofi, A.L. Probing the Flexibility of Zika Virus Envelope Protein DIII Epitopes Using Molecular Dynamics Simulations. Mol. Simul. 2020, 46, 541–547. [Google Scholar] [CrossRef]
- Alaofi, A.L. Exploring Structural Dynamics of the MERS-CoV Receptor DPP4 and Mutant DPP4 Receptors. J. Biomol. Struct. Dyn. 2020, 40, 752–763. [Google Scholar] [CrossRef]
- Alaofi, A.L.; Shahid, M. Mutations of SARS-CoV-2 RBD May Alter Its Molecular Structure to Improve Its Infection Efficiency. Biomolecules 2021, 11, 1273. [Google Scholar] [CrossRef] [PubMed]
- The Glu143 Residue Might Play a Significant Role in T20 Peptide Binding to HIV-1 Receptor Gp41: An In Silico Study—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35745059/ (accessed on 17 September 2022).
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- NCBI Doxorubicin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Doxorubicin (accessed on 3 September 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.; DeLano, W. PyMOL; Schrodinger, LLC: New York, NY, USA, 2020. [Google Scholar]
- Schrödinger Release Maestro; Schrodinger, LLC: New York, NY, USA, 2022.
- Villanueva, R.; Romero-Tamayo, S.; Laplaza, R.; Martínez-Olivan, J.; Velázquez-Campoy, A.; Sancho, J.; Ferreira, P.; Medina, M. Redox- and Ligand Binding-Dependent Conformational Ensembles in the Human Apoptosis-Inducing Factor Regulate Its Pro-Life and Cell Death Functions. Antioxid. Redox Signal. 2019, 30, ars.2018.7658. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-Based Design Identifies a Potent NUPR1 Inhibitor Exerting Anticancer Activity via Necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, J.; Latorre, P.; Carrodeguas, J.A.; Velázquez-Campoy, A.; Sancho, J.; López-Buesa, P. Inhibition of Pig Phosphoenolpyruvate Carboxykinase Isoenzymes by 3-Mercaptopicolinic Acid and Novel Inhibitors. PLoS ONE 2016, 11, e0159002. [Google Scholar] [CrossRef] [Green Version]
- Abian, O.; Vega, S.; Sancho, J.; Velazquez-Campoy, A. Allosteric Inhibitors of the NS3 Protease from the Hepatitis C Virus. PLoS ONE 2013, 8, e69773. [Google Scholar] [CrossRef]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thöny, B.; Sancho, J.; Martinez, A. Identification of Pharmacological Chaperones as Potential Therapeutic Agents to Treat Phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Velázquez-Campoy, A.; Martínez-Júlvez, M.L.; Neira, J.; Pérez-Dorado, I.; Hermoso, J.; Jiménez, P.; Lanas, A.; S. Hoffman, P.; Sancho, J. Discovery of Specific Flavodoxin Inhibitors as Potential Therapeutic Agents against Helicobacter Pylori Infection. ACS Chem. Biol. 2009, 4, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Sancho, J.; Abian, O.; Vega, S. Biophysical Screening for Identifying Pharmacological Chaperones and Inhibitors Against Conformational and Infectious Diseases. Curr. Drug Targets 2016, 17, 1492–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-H.; Chuang, S.-J.; Chen, C.-C.; Cheng, S.-C.; Cheng, K.-W.; Lin, C.-H.; Sun, C.-Y.; Chou, C.-Y. Structural and Functional Characterization of MERS Coronavirus Papain-like Protease. J. Biomed. Sci. 2014, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, S.J.F.; De Burghgraeve, T.; Froeyen, M.; Pastorino, B.; Alen, M.M.F.; Mondotte, J.A.; Herdewijn, P.; Jacobs, M.; De Lamballerie, X.; Schols, D.; et al. A Derivate of the Antibiotic Doxorubicin Is a Selective Inhibitor of Dengue and Yellow Fever Virus Replication in Vitro. Antimicrob. Agents Chemother. 2010, 54, 5269–5280. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaofi, A.L.; Shahid, M.; Raish, M.; Ansari, M.A.; Syed, R.; Kalam, M.A. Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro. Molecules 2022, 27, 7553. https://doi.org/10.3390/molecules27217553
Alaofi AL, Shahid M, Raish M, Ansari MA, Syed R, Kalam MA. Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro. Molecules. 2022; 27(21):7553. https://doi.org/10.3390/molecules27217553
Chicago/Turabian StyleAlaofi, Ahmed L., Mudassar Shahid, Mohammad Raish, Mushtaq Ahmad Ansari, Rabbani Syed, and Mohd Abul Kalam. 2022. "Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro" Molecules 27, no. 21: 7553. https://doi.org/10.3390/molecules27217553
APA StyleAlaofi, A. L., Shahid, M., Raish, M., Ansari, M. A., Syed, R., & Kalam, M. A. (2022). Identification of Doxorubicin as Repurposing Inhibitory Drug for MERS-CoV PLpro. Molecules, 27(21), 7553. https://doi.org/10.3390/molecules27217553