

The Influence of the Halide in the Crystal Structures of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Halides

Abstract
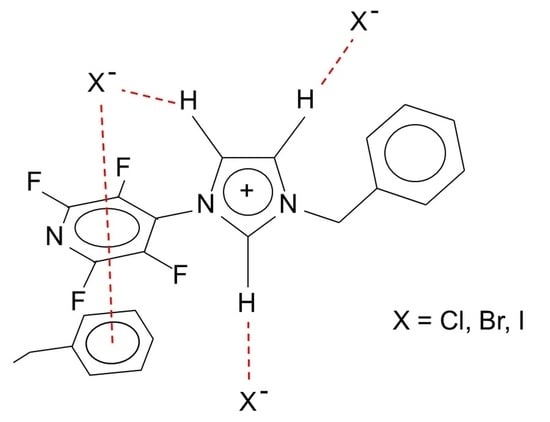
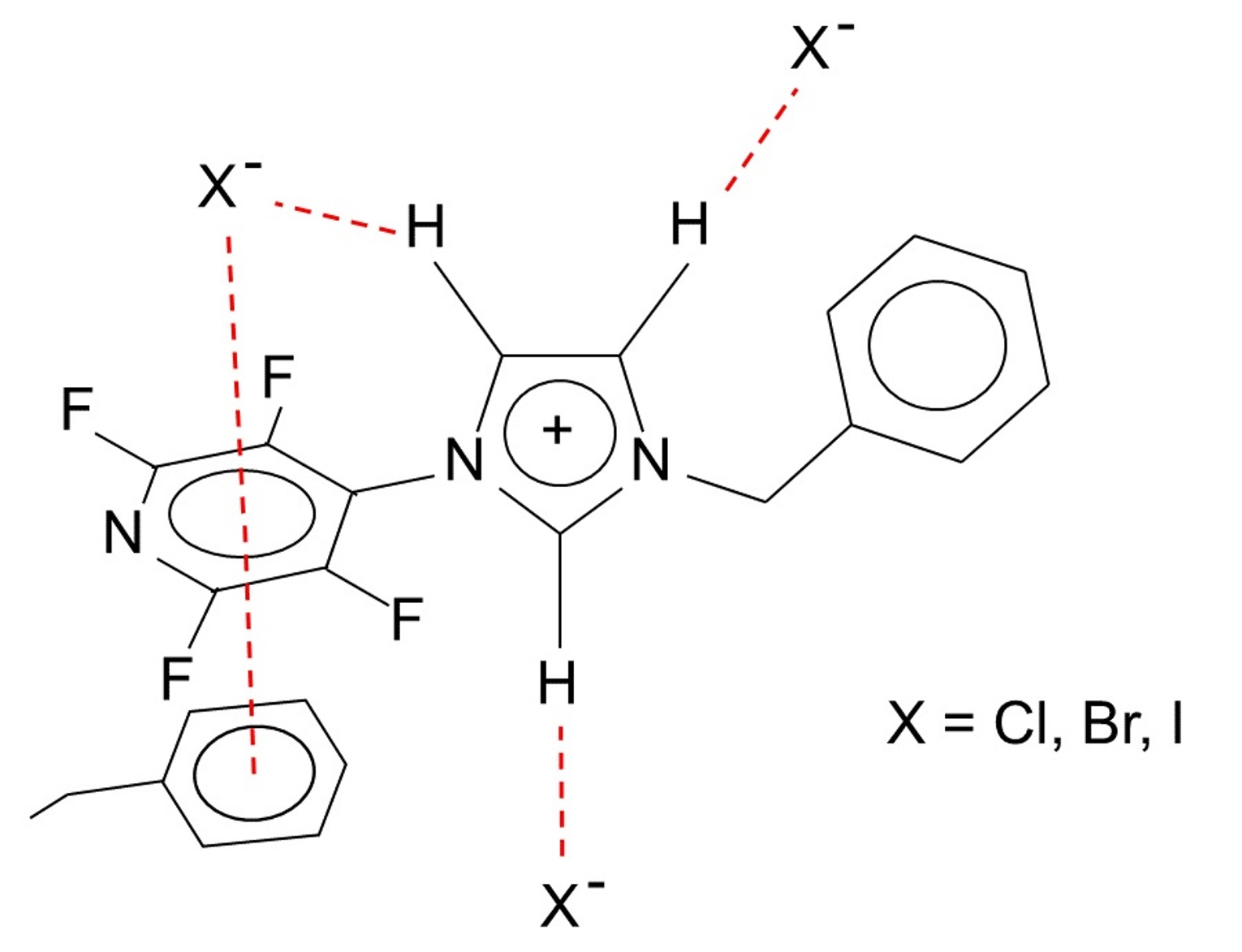
1. Introduction
 1 X = Cl, 2 X = Br, 3 X = I.
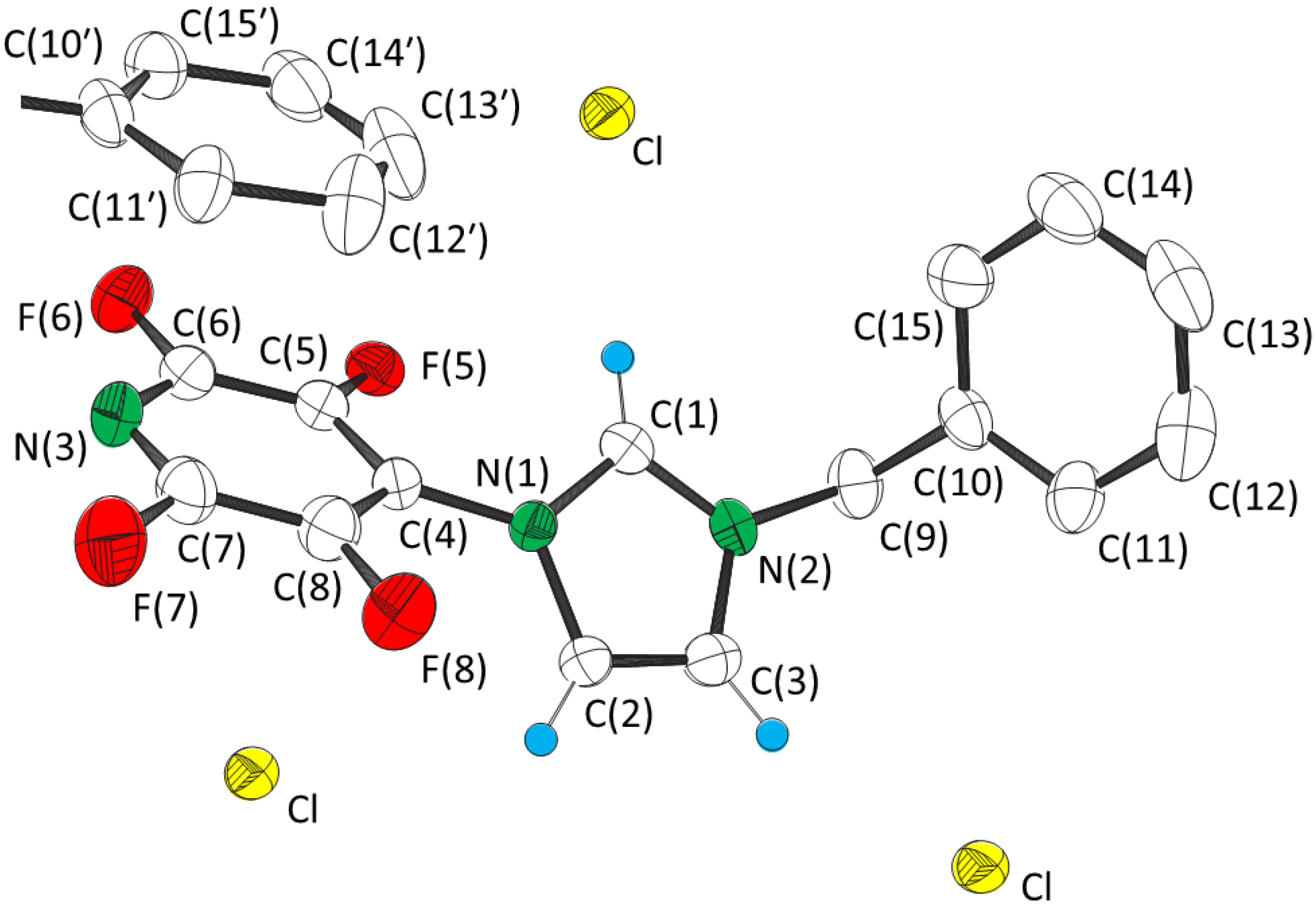
1 X = Cl, 2 X = Br, 3 X = I.2. Results and Discussion
3. Materials and Methods
3.1. Instrumentation
3.2. Materials
3.3. Preparation of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Chloride (1) and Iodide (3)
- 1.
- MS (positive ion): C15H10F4N3 requires 308.0811; found [M − Cl]+ 308.0653. C30H20F8N635Cl requires 651.1310; found [2M − Cl]+ 651.0992. MS (negative ion): C15H10F4N335Cl2 requires 378.0188; found [M + Cl]− 378.0516. C30H20F8N635Cl3 requires 723.0784; found [2M + Cl]− 723.1200.
- 3.
- MS (positive ion): C15H10F4N3 requires 308.0811; found [M − I]+ 308.1033. C30H20F8N6I requires 743.0666; found [2M − I]+ 743.1140. MS (negative ion): I requires 126.9045; found [M − C15H10F4N3]− 126.9181. C15H10F4N3I2 requires 561.8900; found [M + I]− 561.9393.
3.4. X-ray Crystallography
3.5. Density Functional Theory Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Arcus, V.L.; Bernstein, D.R.; Crombie, C.W.; Saunders, G.C. Infinite stacking of alternating polyfluoroaryl rings and bromide anions. CrystEngComm 2013, 15, 9841–9843. [Google Scholar] [CrossRef]
- Oosterwijk, M.A.; Saunders, G.C.; Zou, W. Polyfluoroarene–arene π–π stacking in three directions to provide crystal polarity. J. Fluor. Chem. 2016, 188, 80–84. [Google Scholar] [CrossRef]
- Althagbi, H.I.; Edwards, A.J.; Nicholson, B.K.; Reason, D.A.; Saunders, G.C.; Sim, S.A.; van der Heijden, D.A. Arene–perfluoroarene–anion stacking and hydrogen bonding interactions in imidazolium salts for the crystal engineering of polarity. Cryst. Growth Des. 2016, 16, 174–188. [Google Scholar] [CrossRef]
- Saunders, G.C.; Thomas, H.P. Crystal structure and theoretical calculations of 1-(4-trifluoromethyl-2,3,5,6-tetrafluorophenyl)-3-benzylimidazolium bromide. J. Struct. Chem. 2017, 58, 211–215. [Google Scholar] [CrossRef]
- Althagbi, H.I.; Evans, D.N.; Saunders, G.C. The crystal structures of 1-(4-bromo-2,3,5,6-tetrafluorophenyl)-3-benzylmethylmidazolium bromides. J. Mol. Struct. 2018, 1171, 755–761. [Google Scholar] [CrossRef]
- Althagbi, H.I.; Bernstein, D.R.; Crombie, W.C.; Lane, J.R.; McQuiston, D.K.; Oosterwijk, M.A.; Saunders, G.C.; Zou, W. The crystal structures of 1-(4-halo-2,3,5,6-tetrafluorophenyl)-3-benzylmethylmidazolium bromides: The relative importance of anion–π, lone pair–π, π–π stacking and halogen bonding interactions. J. Fluorine Chem. 2018, 206, 61–71. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC technical report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Tokuda, H.; Mikami, M. Theoretical analysis of the hydrogen bond of imidazolium C2–H with anions. Phys. Chem. Chem. Phys. 2007, 9, 4780–4784. [Google Scholar] [CrossRef]
- Hori, A. Arene-perfluoroarene interactions in coordination architectures. In The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp. 163–185. [Google Scholar]
- Singh, S.K.; Das, A. The n → π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys. 2015, 17, 9596–9612. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Jaswal, S.S.; Sharma, T.P. Electronic polarisabilities of ions in alkali halide crystals. J. Phys. Chem. Solids 1973, 34, 509–511. [Google Scholar] [CrossRef]
- Leclercq, L.; Simard, M.; Schmitzer, A.R. 1,3-Dibenzylimidazolium salts: A paradigm of water and anion effect on the supramolecular H-bonds network. J. Mol. Struct. 2009, 918, 101–107. [Google Scholar] [CrossRef]
- Leclercq, L.; Schmitzer, A.R. Dibenzylimidazolium halides: From complex molecular network in solid state to simple dimer in solution and gas phase. J. Phys. Chem. A 2008, 112, 4996–5001. [Google Scholar] [CrossRef]
- Saunders, G.C. On the importance of π–π stacking and cation–anion interactions in the construction of non-centrosymmetric networks of bromide salts of imidazolium cations bearing arene and polyfluoroarene rings. CrystEngComm 2011, 13, 1801–1803. [Google Scholar] [CrossRef][Green Version]
- Mardirossian, N.; Head-Gordon, M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular nonbonded contact distances in organic crystal structures: Comparison with distances expected from van der Waals radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Crystallogr. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–556. [Google Scholar] [CrossRef]
- Stammler, H.-G.; Vishnevskiy, Y.V.; Sicking, C.; Mitzel, N.W. Charge density studies on 2,3,5,6-tetrafluoro- and pentafluoropyridine. CrystEngComm 2013, 15, 3536–3546. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 3 | |
|---|---|---|
| Formula | C15H10ClF4N3 | C15H10IF4N3 |
| Formula weight | 343.71 | 435.16 |
| Crystal system | orthorhombic | monoclinic |
| Space group | Pna21 | P21/c |
| a, Å | 13.3971(6) | 7.24395(17) |
| b, Å | 8.8393(4) | 10.4696(3) |
| c, Å | 12.5132(5) | 20.5728(5) |
| β, o | 90 | 93.535(2) |
| V, Å3 | 1481.83(11) | 1557.30(7) |
| Z | 4 | 4 |
| Dc (g cm−3) | 1.541 | 1.856 |
| Crystal size (mm3) | 0.126 × 0.075 × 0.024 | 0.201 × 0.084 × 0.063 |
| μ (mm−1) | 2.728 | 16.587 |
| θ range (°) | 3.00→74.19 | 4.31→73.90 |
| Total reflections | 4568 | 9013 |
| Unique reflections (Rint) | 2400(0.0269) | 3056(0.0293) |
| Observed reflections [I > 2 (I)] | 2155 | 2796 |
| Parameters | 208 | 208 |
| Final R indices [I > 2σ(I)] | R1 = 0.0394, | R1 = 0.0240, |
| wR2 = 0.1173 | wR2 = 0.0600 | |
| R indices (all data) | R1 = 0.0493, | R1 = 0.0281, |
| wR2 = 0.1350 | wR2 = 0.0629 | |
| Weighting scheme | w = 1/[σ2(Fo)2 + {0.1002 (Fo2 + 2Fc2)/3}2] | w = 1/[σ2(Fo)2 + {0.0352 (Fo2 + 2Fc2)/3}2 + 0.5871(Fo2 + 2Fc2)/3] |
| Max., min. Δρ (eÅ−3) | 0.396, −0.340 | 0.566, −0.773 |
| Goodness of fit on F2 | 1.055 | 1.054 |
| Flack parameter | −0.03(2) | ― |
| 1 | 2 [2] | 3 | Calc 2 | |
|---|---|---|---|---|
| C(1)–N(1) | 1.337(6) | 1.342(2) | 1.346(3) | 1.341 |
| C(1)–N(2) | 1.323(5) | 1.313(2) | 1.324(4) | 1.322 |
| N(1)–C(2) | 1.397(4) | 1.393(2) | 1.391(3) | 1.389 |
| N(1)–C(4) | 1.406(5) | 1.416(2) | 1.413(3) | 1.423 |
| N(2)–C(3) | 1.385(4) | 1.385(2) | 1.380(4) | 1.383 |
| N(2)–C(9) | 1.485(5) | 1.485(2) | 1.469(3) | 1.489 |
| C(2)–C(3) | 1.340(6) | 1.348(3) | 1.345(4) | 1.353 |
| C(9)–C(10) | 1.498(5) | 1.511(3) | 1.512(4) | 1.506 |
| N(1)–C(1)–N(2) | 107.7(3) | 107.7(2) | 107.9(2) | 108.6 |
| C(1)–N(1)–C(2) | 108.6(3) | 109.2(2) | 108.9(2) | 108.5 |
| N(1)–C(2)–C(3) | 106.9(3) | 106.2(2) | 106.3(3) | 106.7 |
| N(2)–C(3)–C(2) | 107.0(3) | 107.1(2) | 107.8(2) | 107.3 |
| C(1)–N(2)–C(3) | 109.8(3) | 109.9(2) | 109.2(2) | 108.9 |
| C(1)–N(1)–C(4) | 125.7(3) | 125.1(2) | 124.4(2) | 125.3 |
| C(1)–N(2)–C(9) | 125.9(3) | 125.4(2) | 124.2(2) | 125.5 |
| N(2)–C(9)–C(10) | 112.8(3) | 110.7(2) | 111.9(2) | 111.1 |
| ∠ C5F4Nplane C3N2plane 3 | 42.1(5) | 39.4(3) | 52.3(5) | 47.0 |
| ∠ C6H5plane C3N2plane 3 | 67.1(5) | 72.3(3) | 86.5(5) | 89.3 |
| ∠ C5F4Nplane C6H5plane 3 | 89.3(5) | 89.8(3) | 39.0(5) | 50.2 |
| C(1)–N(1)–C(4)–C(5) | 40.2(5) | −144.3(2) | −51.8(4) | 46.9 |
| C(1)–N(1)–C(4)–C(8) | −143.7(4) | 39.7(2) | 125.7(3) | −133.6 |
| C(1)–N(2)–C(9)–C(10) | 84.5(4) | 98.1(2) | −97.3(3) | 140.0 |
| C(3)–N(2)–C(9)–C(10) | −95.8(4) | −80.2(2) | 76.2(3) | −41.8 |
| N(2)–C(9)–C(10)–C(11) | 92.9(4) | 81.7(2) | 23.2(3) | 106.6 |
| N(2)–C(9)–C(10)–C(15) | −87.0(4) | −96.1(2) | −157.1(2) | −72.6 |
| 1 (X = Cl) | 2 (X = Br) | 3 (X = I) | ||||
|---|---|---|---|---|---|---|
| Expt | Calc 2 | Expt | Calc 2 | Expt | Calc 2 | |
| C(1)···X− | 3.356(4) | 3.033 | 3.467(2) | 3.169 | 3.576(3) 3.705(3) | 3.198 3.351 |
| N(1)–C(1)···X− | 125.6(2) | 140.9 | 135.0(1) | 132.6 | 93.3(2) 136.9(2) | 91.2 144.2 |
| N(2)–C(1)···X− | 126.2(2) | 110.2 | 117.3(1) | 119.7 | 136.2(2) 97.8(2) | 127.3 106.7 |
| C3N2plane···X− 3 | 0.357(4) | 0.489) | 0.15(1) | 0.10 | 2.288(3) 2.130(3) | 2.427 0.438 |
| C5F4Nplane···X− 3 | – | – | – | – | 3.596(4) | 3.357 |
| C5F4N+···X− 4 | – | – | – | – | 3.815(4) | 3.836 |
| C(4)···X− | – | – | – | – | 3.604(3) | 3.414 |
| C(2)···X− | 3.499(4) | 2.917 | 3.834(2) | 3.069 | 3.973(3) | 3.367 |
| N(1)–C(2)···X− | 87.0(2) | 82.2 | 83.66(9) | 82.7 | 84.1(2) | 85.8 |
| C(3)–C(2)···X− | 141.0(3) | 120.8 | 141.0(1) | 122.1 | 138.9(2) | 139.5 |
| C3N2plane···X− 3 | 2.218(4) | 2.518 | 2.388(2) | 2.605 | 2.577(3) | 2.154 |
| C5F4Nplane···X− 3 | 3.192(4) | 2.967 | 3.315(2) | 3.111 | 3.631(4) | 3.285 |
| C5F4N+···X− 4 | 3.372(4) | 3.518 | 3.414(2) | 3.599 | 3.847(4) | 3.634 |
| C(4)···X− | 3.289(4) | 3.024 | 3.441(2) | 3.146 | 3.671(3) | 3.292 |
| C(3)··· X− | 3.449(4) | 3.134 | 3.593(2) | 3.285 | – | – |
| N(2)–C(3)···X− | 110.3(2) | 101.3 | 109.7(1) | 101.4 | – | – |
| C(2)–C(3)···X− | 142.6(3) | 150.9 | 139.6(1) | 145.6 | – | – |
| C3N2plane···X− 3 | 0.140(4) | 0.358 | 0.97(1) | 1.061 | – | – |
| C(3)··· X− | 3.449(4) | 3.134 | 3.593(2) | 3.285 | – | – |
| ∠ C6H5plane C5F4N plane 3 | 2.6(3) | – | 4.8(2) | – | – | – |
| C6H5+···C5F4N plane 3 | 3.322(6) | – | 3.271(3) | – | – | – |
| C6H5plane···C5F4N+ 3,4 | 3.351(6) | – | 3.374(3) | – | – | – |
| C6H5+···C5F4N+ 4 | 3.499(6) | – | 3.614(3) | – | – | – |
| C5F4N plane···C5F4N plane 3,5 | – | – | – | – | 7.244(4) | – |
| I−···I− 6 | – | – | – | – | 7.2439(3) | – |
| I−··· C5F4N+···I− 7 | – | – | – | – | 142.0(1) | – |
| ∠ column C5F4N plane 8 | – | – | – | – | 86.0(2) | – |
| Halide Close to: | Salt | Experimental Structure | Optimized Halide Position 2 | Electrostatic Interaction (r, Å) 3 | Tetrafluoropyridylimidazole 4 | Pentafluoropyridine 4,5 |
|---|---|---|---|---|---|---|
| C(1) | 1 | −392 | −406 | −336 (4.138) | −65 | |
| 2 | −374 | −380 | −327 (4.244) | −47 | ||
| 3 | −366 −342 | −371 −354 | −336 (4.134) −322 (4.318) | −68 −20 | −51 | |
| C(2) | 1 | −352 | −369 | −311 (4.466) | −86 | −62 |
| 2 | −332 | −351 | −295 (4.702) | −77 | −56 | |
| 3 | −324 | −334 | −290 (4.790) | −71 | −51 | |
| C(3) | 1 | −362 | −379 | −287 (4.832) | −31 | |
| 2 | −343 | −353 | −281 (4.938) | −22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharige, U.A.I.; Saunders, G.C. The Influence of the Halide in the Crystal Structures of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Halides. Molecules 2022, 27, 7634. https://doi.org/10.3390/molecules27217634
Acharige UAI, Saunders GC. The Influence of the Halide in the Crystal Structures of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Halides. Molecules. 2022; 27(21):7634. https://doi.org/10.3390/molecules27217634
Chicago/Turabian StyleAcharige, Udari A. I., and Graham C. Saunders. 2022. "The Influence of the Halide in the Crystal Structures of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Halides" Molecules 27, no. 21: 7634. https://doi.org/10.3390/molecules27217634
APA StyleAcharige, U. A. I., & Saunders, G. C. (2022). The Influence of the Halide in the Crystal Structures of 1-(2,3,5,6-Tetrafluoro-4-pyridyl)-3-benzylimidazolium Halides. Molecules, 27(21), 7634. https://doi.org/10.3390/molecules27217634