
Structural Homology-Based Drug Repurposing Approach for Targeting NSP12 SARS-CoV-2

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
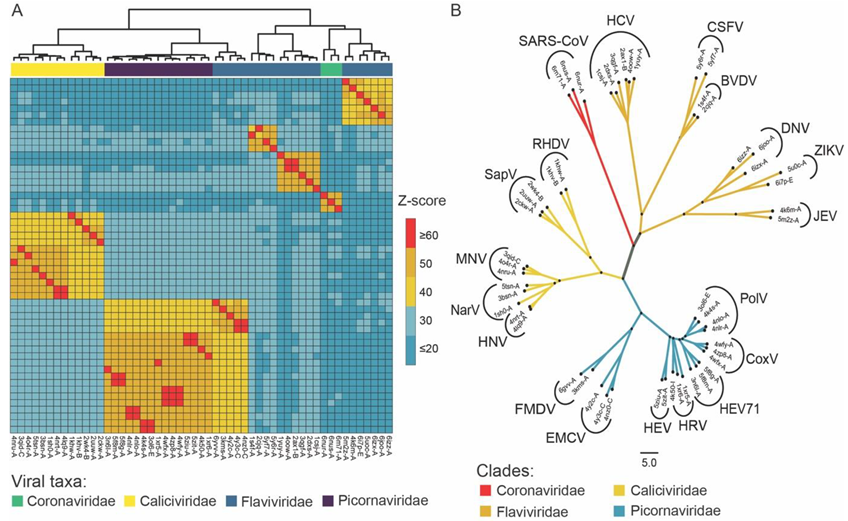
2.1. Structural Similarity-Based Searching and Screening for Homologous Proteins
2.2. Sequence and Structural Analysis of Conserved Motifs in NSP-12
2.3. Pharmacophore Modeling and Druggable Site in NSP-12
2.4. Molecular Docking of NSP-12 with FDA-Approved Antivirals
2.5. Molecular Dynamics (MD) Simulation
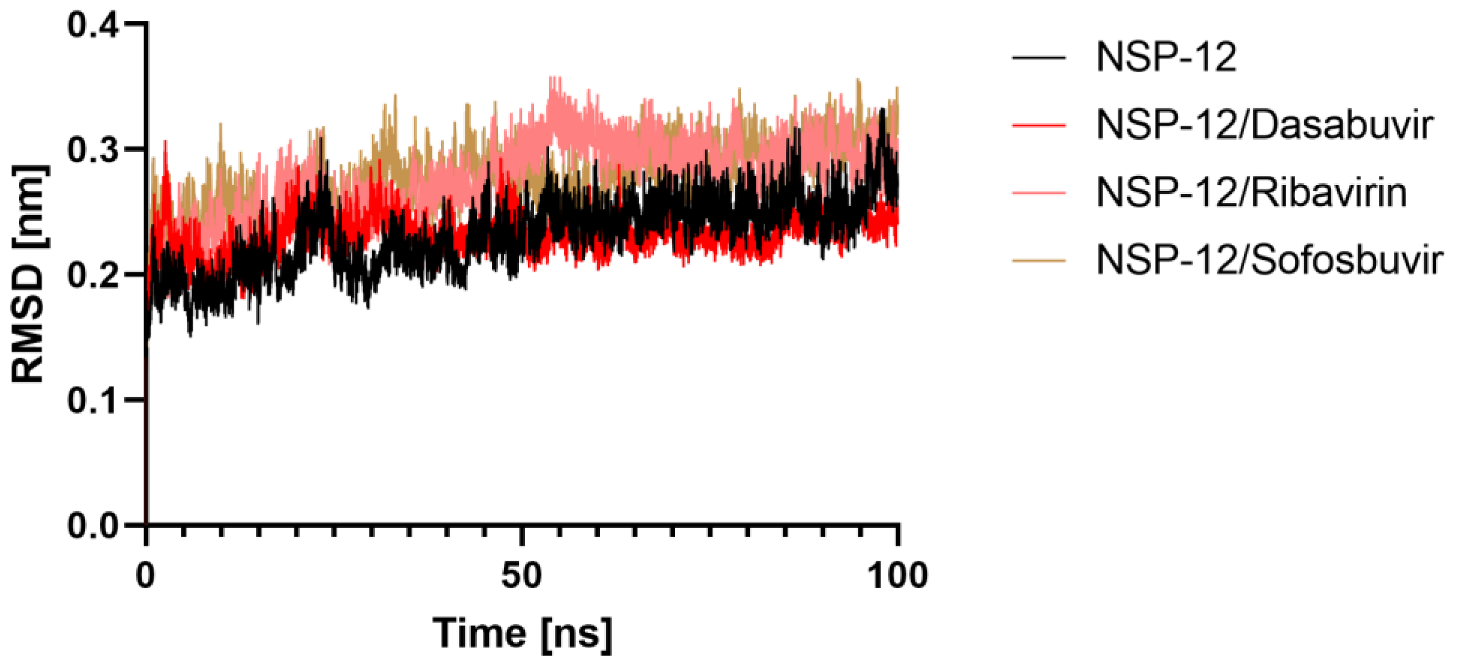
2.5.1. Root Mean Square Deviation (RMSD)
2.5.2. Root Mean Square Fluctuation (RMSF)
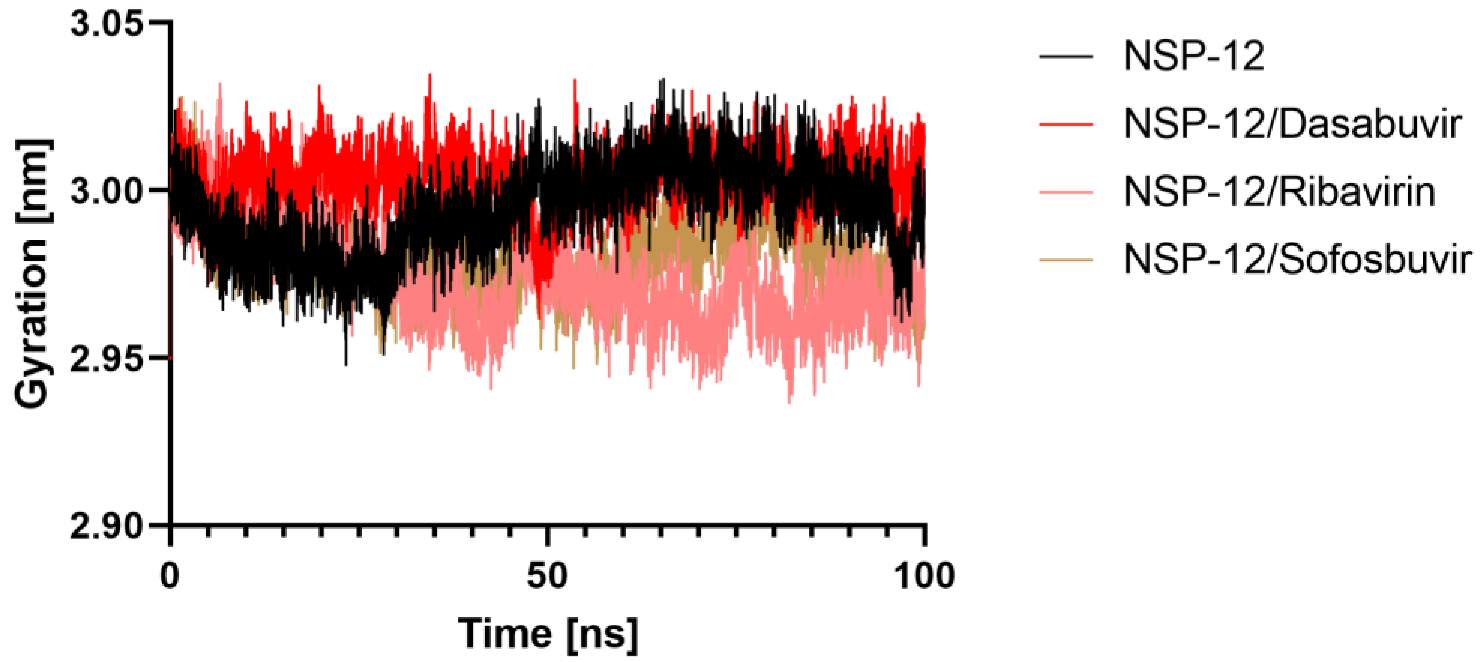
2.5.3. Radius of Gyration
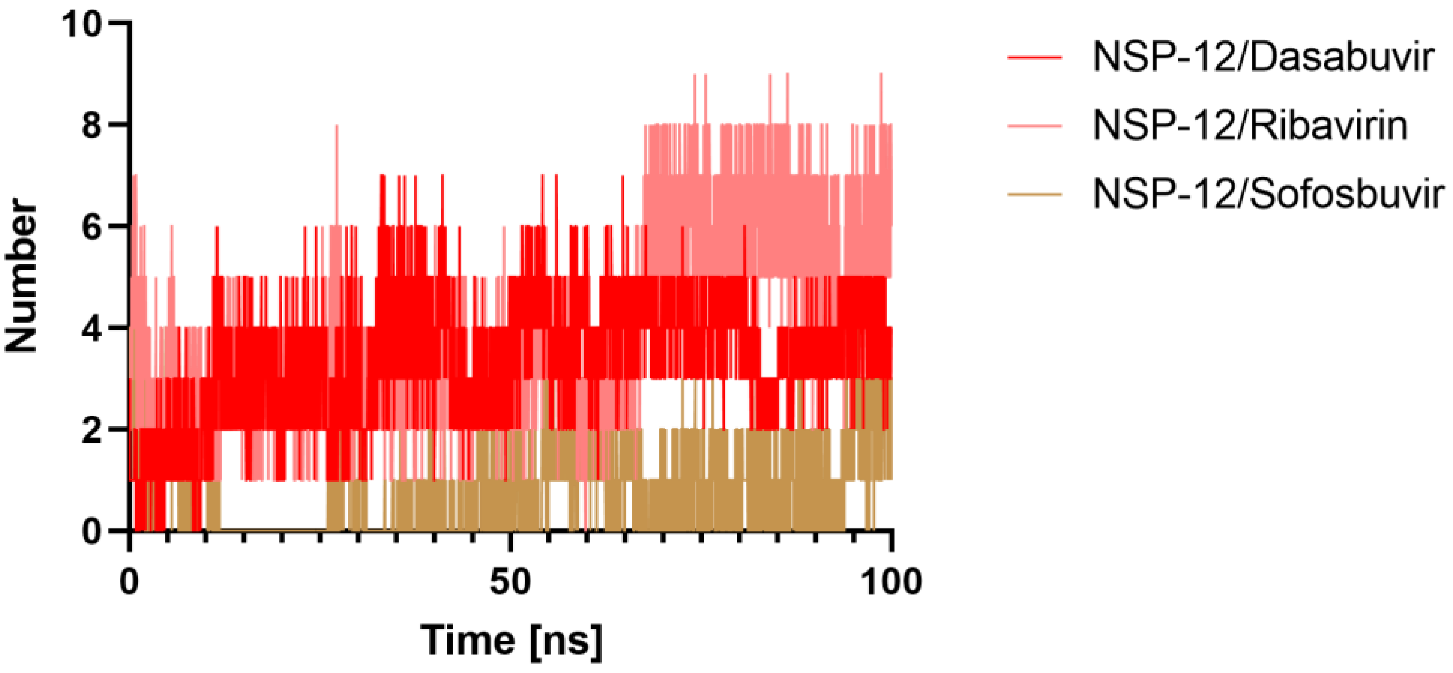
2.5.4. Hydrogen Bonds Analysis
2.6. MMPBSA Binding Energy
3. Discussion
4. Material and Methods
4.1. Structural-Based Search for Homologous Proteins
4.2. Homologous Structures and Amino Acid Sequence Analysis
4.3. Pharmacophore Modeling and Prediction of Druggable Sites in NSP-12
4.4. Preparation of Ligand and Receptor
4.5. Molecular Docking
4.6. Molecular Dynamics (MD) Simulation
4.7. MM-PBSA Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeu, Y.; Yoon, Y.; Park, S. Protein localization vector propagation: A method for improving the accuracy of drug repositioning. Mol. BioSyst. 2015, 11, 2096–2102. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/news-room/detail/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 7 May 2020).
- McKibbin, W.; Fernando, R. The Global Macroeconomic Impacts of COVID-19: Seven Scenarios; CAMA Working Paper No. 19/2020; SSRN: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 29 May 2020).
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus infections—More than just the common cold. JAMA 2020, 323, 707–708. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J.M. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.M.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- Brian, D.; Baric, R. Coronavirus genome structure and replication. In Coronavirus Replication and Reverse Genetics; Springer: Germany, 2005; pp. 1–30. [Google Scholar]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.C.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.K.-S.; Arnold, J.J.; Cameron, C.E. Structure-function relationships among RNA-dependent RNA polymerases. RNA Interf. 2008, 320, 137–156. [Google Scholar] [CrossRef]
- Choi, K.H. Viral polymerases. In Viral Molecular Machines; Springer: Germany, 2012; pp. 267–304. [Google Scholar]
- Ko, W.-C.; Rolain, J.-M.; Lee, N.-Y.; Chen, P.-L.; Huang, C.-T.; Lee, P.-I.; Hsueh, P.-R. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int. J. Antimicrob. Agents 2020, 55, 105933. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar] [CrossRef]
- Ren, P.X.; Shang, W.J.; Yin, W.C.; Ge, H.; Wang, L.; Zhang, X.L.; Li, B.Q.; Li, H.L.; Xu, Y.C.; Xu, E.H.; et al. A multi-targeting drug design strategy for identifying potent anti-SARS-CoV-2 inhibitors. Acta Pharmacol. Sin. 2022, 43, 483–493. [Google Scholar] [CrossRef]
- Elkarhat, Z.; Charoute, H.; Elkhattabi, L.; Barakat, A.; Rouba, H. Potential inhibitors of SARS-CoV-2 RNA dependent RNA polymerase protein: Molecular docking, molecular dynamics simulations and MM-PBSA analyses. J. Biomol. Struct. Dyn. 2020, 40, 361–374. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, P.P.; Upadhyay, C.; Kempaiah, P.; Rathi, B. Multi-targeting approach for nsp3, nsp9, nsp12 and nsp15 proteins of SARS-CoV-2 by Diosmin as illustrated by molecular docking and molecular dynamics simulation methodologies. Methods 2021, 195, 44–56. [Google Scholar] [CrossRef]
- Singh, S.K.; Upadhyay, A.K.; Reddy, M.S.J.B. Screening of potent drug inhibitors against SARS-CoV-2 RNA polymerase: An in silico approach. 3 Biotech 2021, 11, 93. [Google Scholar] [CrossRef]
- Gentile, I.; Buonomo, A.R.; Borgia, G. Dasabuvir: A non-nucleoside inhibitor of NS5B for the treatment of hepatitis C virus infection. Rev. Recent Clin. Trials 2014, 9, 115–123. [Google Scholar] [CrossRef]
- Feng, J.Y.; Ray, A.S. HCV RdRp, sofosbuvir and beyond. Enzymes 2021, 49, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.J.; Grimes, J.M.; Makeyev, E.V.; Bamford, D.H.; Stuart, D.I. A mechanism for initiating RNA-dependent RNA polymerization. Nature 2001, 410, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef] [PubMed]
- E Gorbalenya, A.; Pringle, F.M.; Zeddam, J.-L.; Luke, B.T.; E Cameron, C.; Kalmakoff, J.; Hanzlik, T.N.; Gordon, K.H.; Ward, V.K. The Palm Subdomain-based Active Site is Internally Permuted in Viral RNA-dependent RNA Polymerases of an Ancient Lineage. J. Mol. Biol. 2002, 324, 47–62. [Google Scholar] [CrossRef]
- Langer, T.; Hoffmann, R.D. Pharmacophore modelling: Applications in drug discovery. Expert Opin. Drug Discov. 2006, 1, 261–267. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe COVID-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- De Albuquerque, P.P.; Santos, L.H.; Antunes, D.; Caffarena, E.R.; Figueiredo, A.S. Structural insights into NS5B protein of novel equine hepaciviruses and pegiviruses complexed with polymerase inhibitors. Virus Res. 2020, 278, 197867. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA dependent RNA polymerases: Insights from structure, function and evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Jia, H.; Gong, P. A structure-function diversity survey of the RNA-dependent RNA polymerases from the positive-strand RNA viruses. Front. Microbiol. 2019, 10, 1945. [Google Scholar] [CrossRef]
- Shu, B.; Gong, P. Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc. Natl. Acad. Sci. USA 2016, 113, E4005–E4014. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali server update. Nucliec Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 13 May 2020).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Hill, A.D.; Reilly, P.J. Scoring functions for AutoDock. Glycoinformatics 2015, 1273, 467–474. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Binding Energy (KJ/mol) | SASA Energy (KJ/mol) | Polar Solvation Energy (KJ/mol) | Electrostatic Energy (KJ/mol) | Van der Waal Energy (KJ/mol) |
|---|---|---|---|---|---|
| NSP-12/Dasabuvir | −42.151 ± 21.735 | −20.615 ± 1.635 | 228.354 ± 25.854 | −87.631 ± 20.339 | −162.259 ± 16.736 |
| NSP-12/Ribavirin | 60.285 ± 28.431 | −8.846 ± 1.638 | 420.099 ± 75.663 | −327.013 ± 76.448 | −23.956 ± 20.920 |
| NSP-12/Sofosbuvir | −26.168 ± 54.225 | −9.734 ± 5.282 | 94.581 ± 68.246 | −26.753 ± 24.025 | −84.262 ± 47.830 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljuaid, A.; Salam, A.; Almehmadi, M.; Baammi, S.; Alshabrmi, F.M.; Allahyani, M.; Al-Zaydi, K.M.; Izmirly, A.M.; Almaghrabi, S.; Baothman, B.K.; et al. Structural Homology-Based Drug Repurposing Approach for Targeting NSP12 SARS-CoV-2. Molecules 2022, 27, 7732. https://doi.org/10.3390/molecules27227732
Aljuaid A, Salam A, Almehmadi M, Baammi S, Alshabrmi FM, Allahyani M, Al-Zaydi KM, Izmirly AM, Almaghrabi S, Baothman BK, et al. Structural Homology-Based Drug Repurposing Approach for Targeting NSP12 SARS-CoV-2. Molecules. 2022; 27(22):7732. https://doi.org/10.3390/molecules27227732
Chicago/Turabian StyleAljuaid, Abdulelah, Abdus Salam, Mazen Almehmadi, Soukayna Baammi, Fahad M. Alshabrmi, Mamdouh Allahyani, Khadijah M. Al-Zaydi, Abdullah M. Izmirly, Sarah Almaghrabi, Bandar K. Baothman, and et al. 2022. "Structural Homology-Based Drug Repurposing Approach for Targeting NSP12 SARS-CoV-2" Molecules 27, no. 22: 7732. https://doi.org/10.3390/molecules27227732
APA StyleAljuaid, A., Salam, A., Almehmadi, M., Baammi, S., Alshabrmi, F. M., Allahyani, M., Al-Zaydi, K. M., Izmirly, A. M., Almaghrabi, S., Baothman, B. K., & Shahab, M. (2022). Structural Homology-Based Drug Repurposing Approach for Targeting NSP12 SARS-CoV-2. Molecules, 27(22), 7732. https://doi.org/10.3390/molecules27227732