

Isolation, Structural Analysis and Biological Activity Assays of Biselisabethoxanes A and B: Two Dissymmetric Bis-Diterpenes from the Southwestern Caribbean Sea Gorgonian Coral Pseudopterogorgia elisabethae
Abstract
:1. Introduction
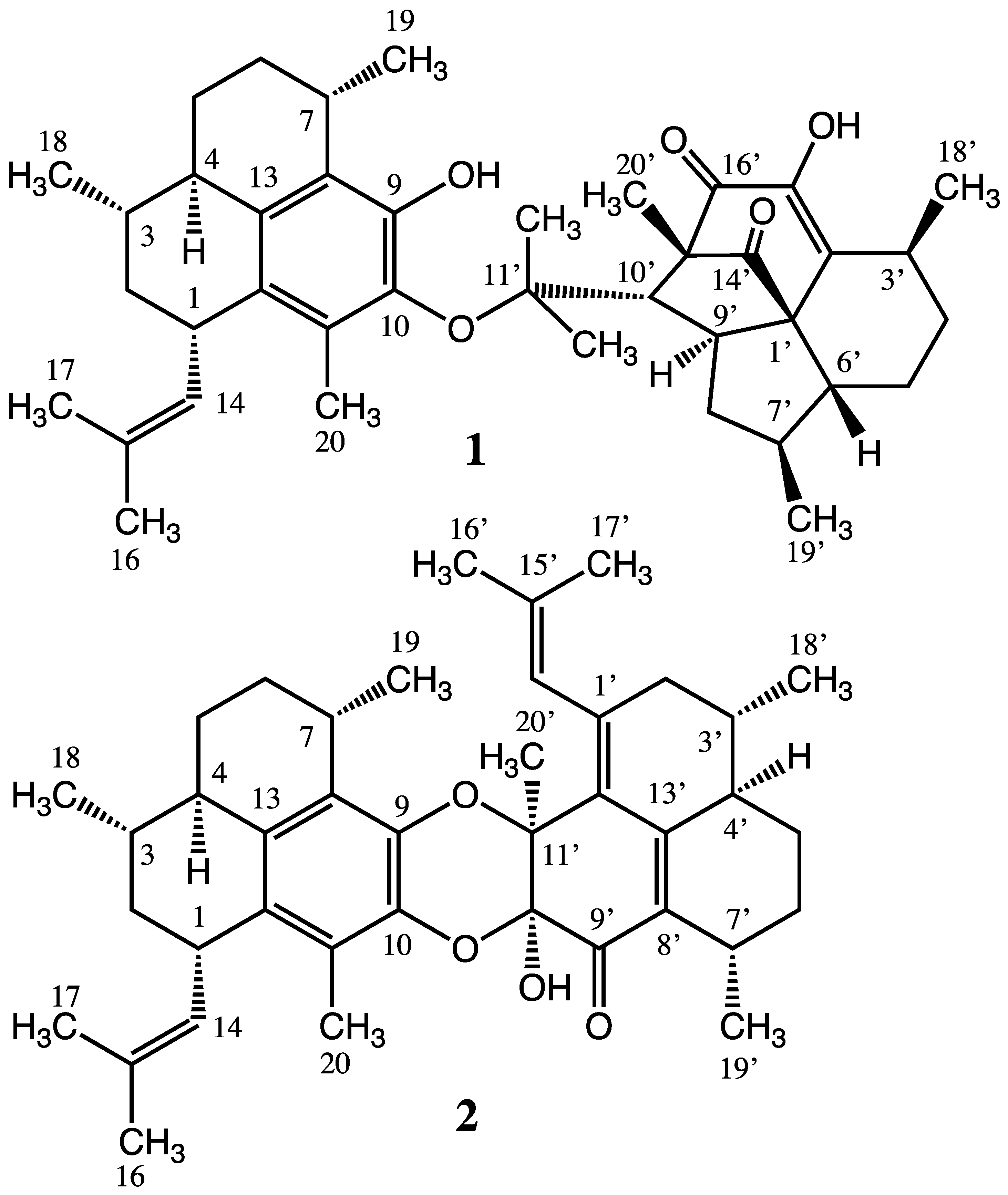
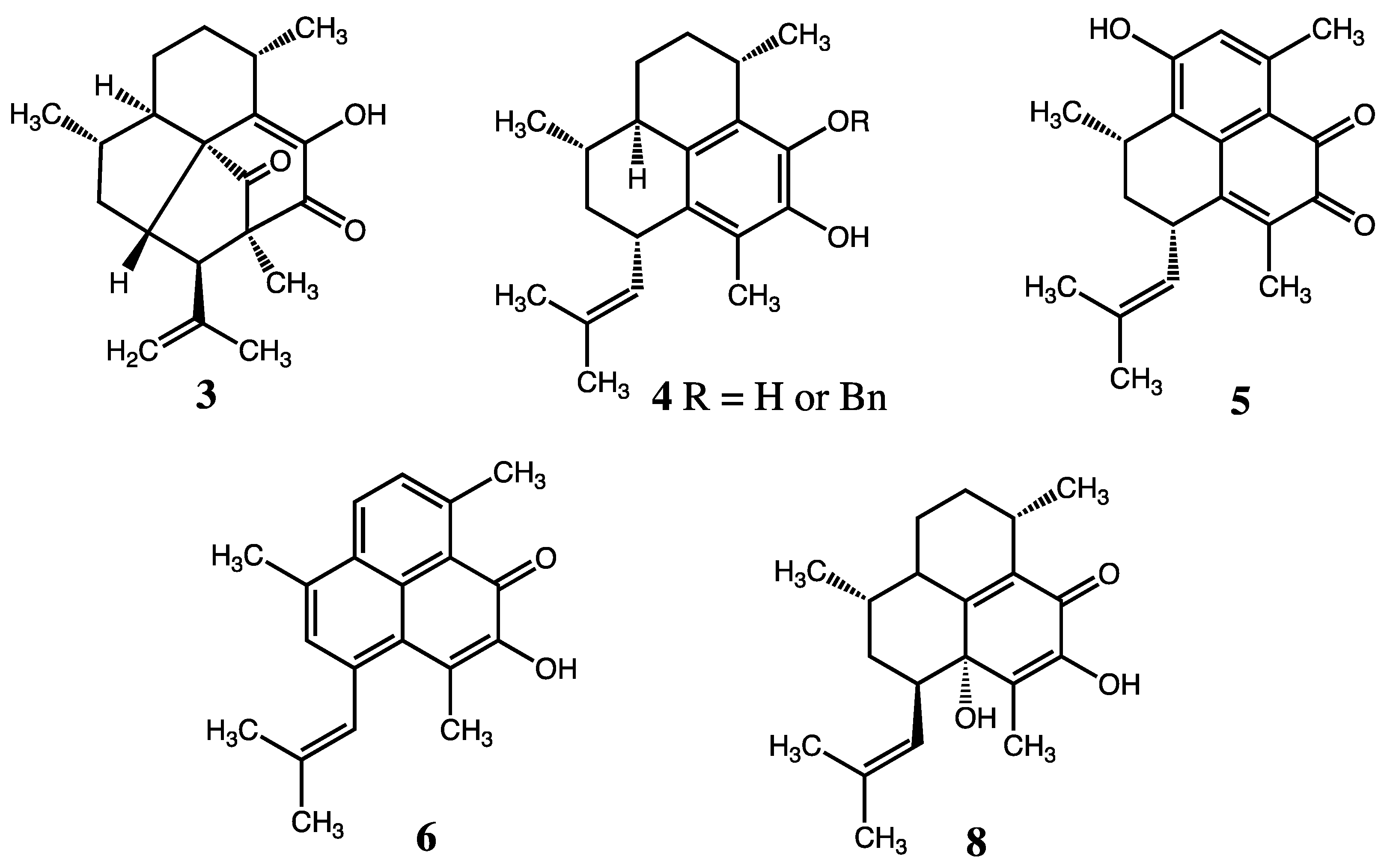
2. Results and Discussion
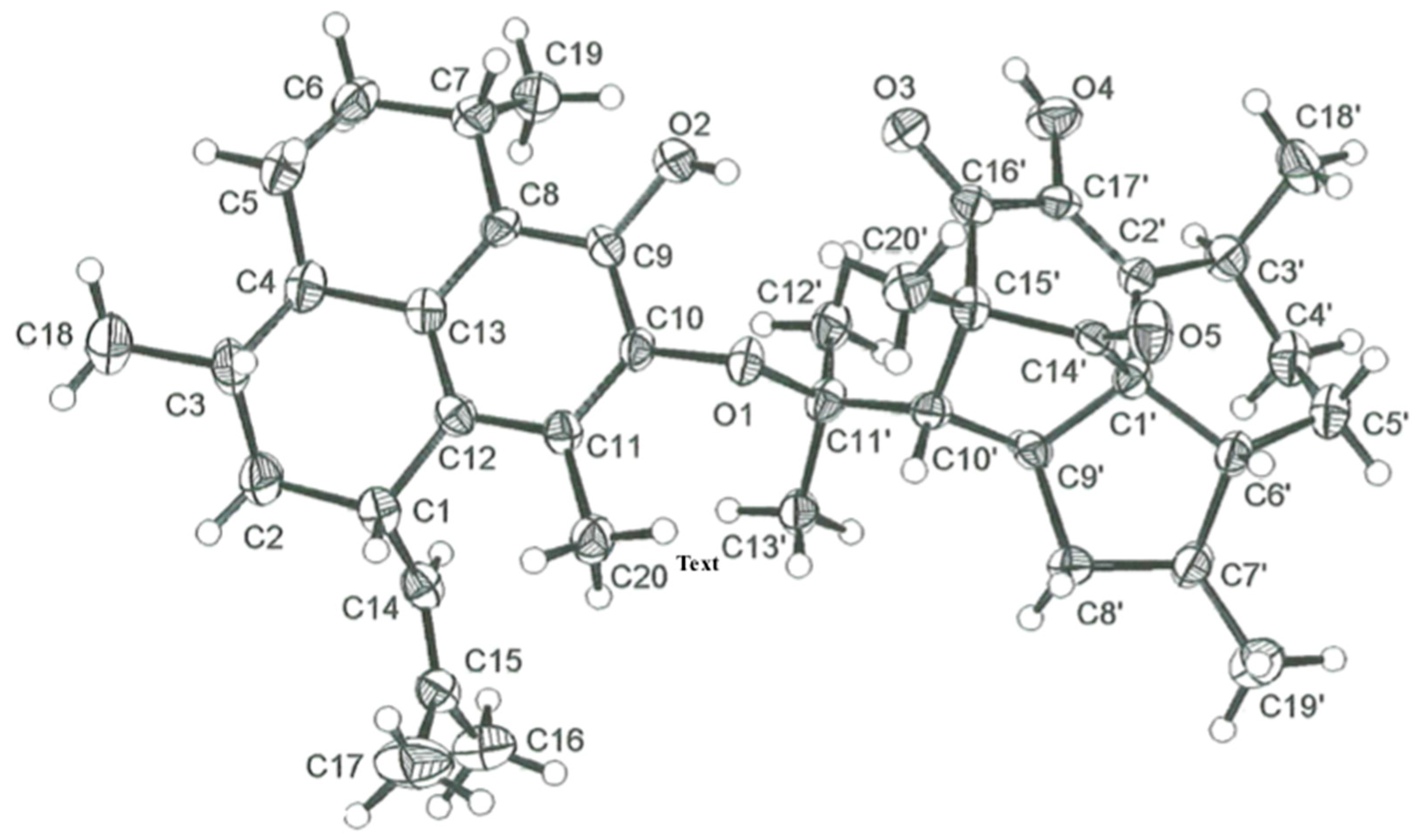
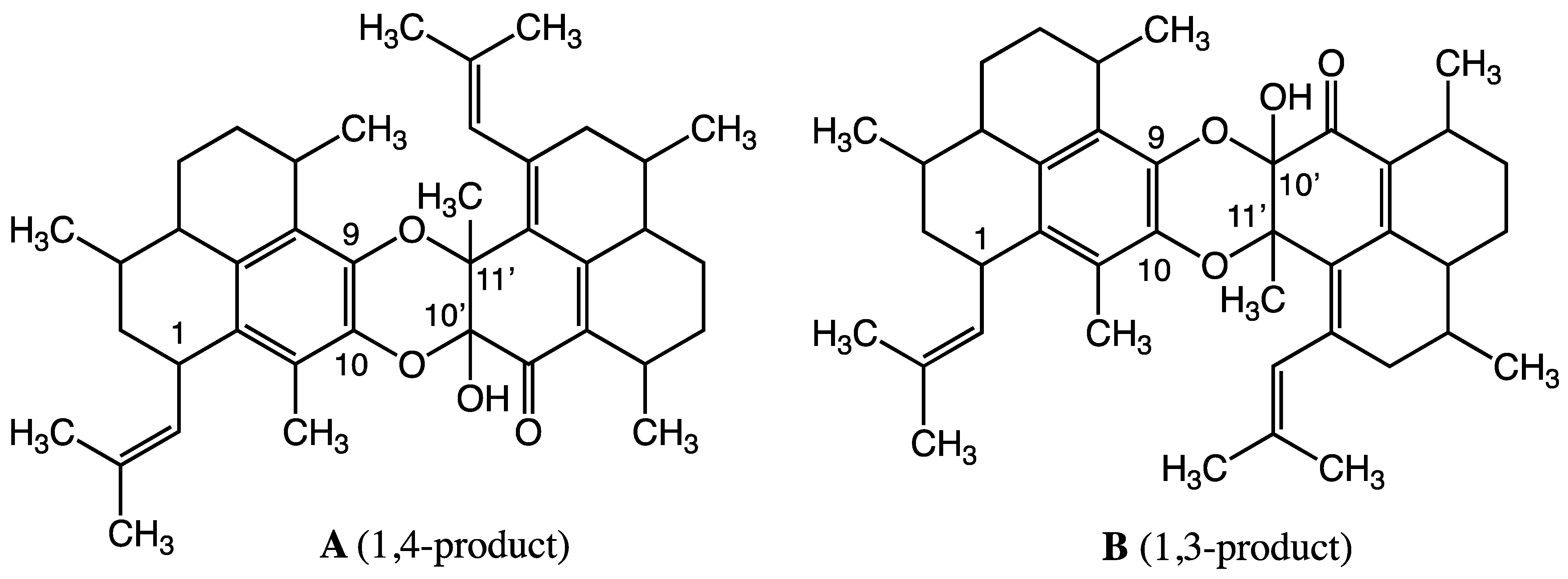
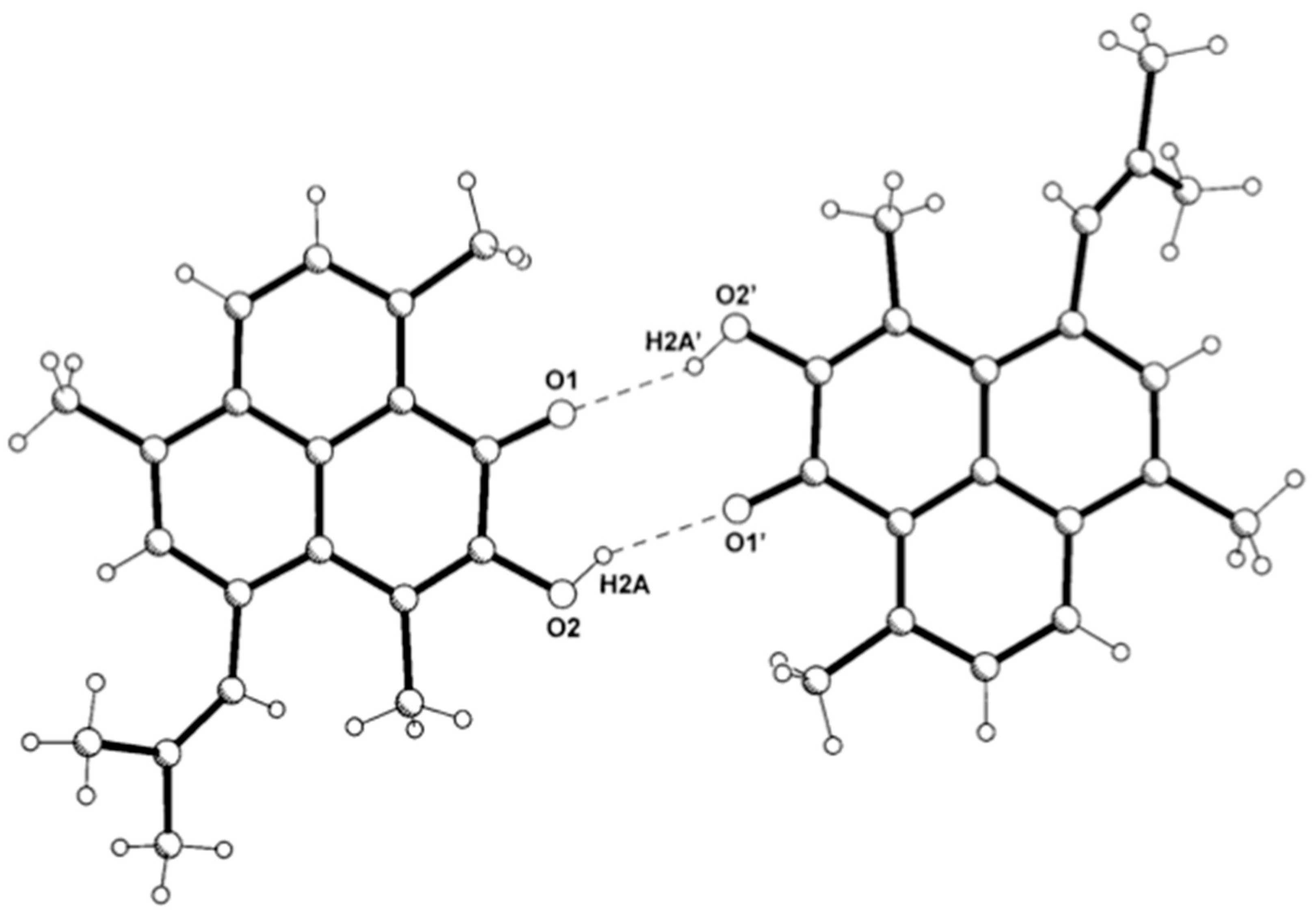
2.1. Structure Elucidation
2.2. Biological Evaluation of the New C40 Bis-Diterpenes
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Collection, Extraction, and Isolation of Bis-Diterpenes 1 and 2
3.4. Biological Assays
3.5. X-ray Crystallographic Data for Biselisabethoxane A (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lin, L.-G.; Ung, C.O.L.; Feng, Z.-L.; Huang, L.; Hu, H. Naturally occurring diterpenoid dimers: Source, biosynthesis, chemistry and bioactivities. Planta Med. 2016, 82, 1309–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.-H.; Elmadfa, I. Biological relevance of terpenoids. Ann. Nutr. Metab. 2003, 47, 95–106. [Google Scholar] [PubMed]
- Avila, C. Terpenoids in marine heterobranch molluscs. Mar. Drugs 2020, 18, 162. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2022, 39, 1122–1171, and Previous Reports in This Series. [Google Scholar] [PubMed]
- Look, S.A.; Fenical, W.; Jacobs, R.S.; Clardy, J. The pseudopterosins: Anti-inflammatory and analgesic natural products from the sea whip Pseudopterogorgia elisabethae. Proc. Natl. Acad. Sci. USA 1986, 83, 6238–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A.M.S.; Jacobson, P.B.; Fenical, W.; Jacobs, R.S.; Glaser, K.B. Pharmacological characterization of the pseudopterosins: Novel anti-inflammatory natural products isolated from the Caribbean soft coral, Pseudopterogorgia elisabethae. Life Sci. 1998, 62, 401–407. [Google Scholar]
- Roussis, V.; Wu, Z.; Fenical, W.; Strobel, S.A.; Van Duyne, G.D.; Clardy, J. New anti-inflammatory pseudopterosins from the marine octocoral Pseudopterogorgia elisabethae. J. Org. Chem. 1990, 55, 4916–4922. [Google Scholar] [CrossRef]
- Look, S.A.; Fenical, W. The seco-pseudopterosins, new anti-inflammatory diterpene-glycosides from a Caribbean gorgonian octocoral of the genus Pseudopterogorgia. Tetrahedron 1987, 43, 3363–3370. [Google Scholar]
- Ferns, T.A.; Kerr, R.G. Identification of amphilectosins as key intermediates in pseudopterosin biosynthesis. J. Org. Chem. 2005, 70, 6152–6157. [Google Scholar]
- Ata, A.; Kerr, R.G.; Moya, C.E.; Jacobs, R.S. Identification of anti-inflammatory diterpenes from the marine gorgonian Pseudopterogorgia elisabethae. Tetrahedron 2003, 59, 4215–4222. [Google Scholar]
- Correa, H.; Aristizabai, F.A.; Duque, C.; Kerr, R.G. Cytotoxic and antimicrobial activity of pseudopterosins and seco-pseudopterosins isolated from the octocoral Pseudopterogorgia elisabethae of San Andrés and Providencia islands (Southwest Caribbean Sea). Mar. Drugs 2011, 9, 334–343. [Google Scholar] [PubMed] [Green Version]
- Brück, T.B.; Kerr, R.G. Purification and kinetic properties of elisabethatriene synthase from the coral Pseudopterogorgia elisabethae. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol. 2006, 143, 269–278. [Google Scholar]
- Duque, C. Pseudopterogorgia elisabethae de San Andrés y Providencia, una pluma de mar con excelente potencial como fuente de productos naturales con aplicación industrial. Rev. Acad. Colomb. Cienc. 2010, 130, 89–103. [Google Scholar]
- Puyana, M.; Narvaez, G.; Paz, A.; Osorno, O.; Duque, C. Pseudopterosin content variability of the purple sea whip Pseudopterogorgia elisabethae at the islands of San Andrés and Providencia (SW Caribbean). J. Chem. Ecol. 2004, 30, 1183–1201. [Google Scholar]
- Correa, H.; Valenzuela, A.L.; Ospina, L.F.; Duque, C. Anti-inflammatory effects of the gorgonian Pseudopterogorgia elisabethae collected at the islands of Providencia and San Andrés (SW Caribbean). J. Inflamm. 2009, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque, C.; Puyana, M.; Narvaez, G.; Paz, A.; Osorno, O.; Hara, N.; Fujimoto, Y. Pseudopterosins P–V, new compounds from the gorgonian octocoral Pseudopterogorgia elisabethae from Providencia island, Colombian Caribbean. Tetrahedron 2004, 60, 10627–10635. [Google Scholar]
- Duque, C.; Puyana, M.; Castellanos, L.; Arias, A.; Correa, H.; Osorno, O.; Asai, T.; Hara, N.; Fujimoto, Y. Further studies on the constituents of the gorgonian octocoral Pseudopterogorgia elisabethae collected in San Andrés and Providencia islands, Colombian Caribbean: Isolation of a putative biosynthetic intermediate leading to erogorgiaene. Tetrahedron 2006, 62, 4205–4213. [Google Scholar] [CrossRef]
- Rodríguez, A.D. The natural products chemistry of West Indian gorgonian octocorals. Tetrahedron 1995, 51, 4571–4618. [Google Scholar]
- Rodríguez, A.D.; Rodríguez, I.I. Isolation and structure of biselisabethoxanes A–E, a rare family of bis-diterpenes from Pseudopterogorgia elisabethae. In Proceedings of the Marine Natural Products Gordon Research Conference, Ventura, CA, USA, 24–29 February 2008. [Google Scholar]
- Rodríguez, A.D.; Rodríguez, I.I. Biselisabethoxanes A–D, an unusual family of bis-diterpenes from the gorgonian coral Pseudopterogorgia elisabethae. In Proceedings of the 61st Southeastern Regional Meeting of the American Chemical Society (SERMACS), San Juan, PR, USA, 21–23 October 2009. Paper BIORG 480. [Google Scholar]
- Yang, B.; Gao, S. Recent advances in the application of Diels–Alder reactions involving o-quinodimethanes, aza-o-quinone methides and o-quinone methides in natural product total synthesis. Chem. Soc. Rev. 2018, 47, 7926–7953. [Google Scholar]
- Nawrat, C.C.; Moody, C.J. Quinones as dienophiles in the Diels–Alder reaction: History and applications in total synthesis. Angew. Chem. Int. Ed. 2014, 53, 2056–2077. [Google Scholar] [CrossRef]
- Look, S.A.; Fenical, W.; Matsumoto, G.K.; Clardy, J. The pseudopterosins: Anti-inflammatory and analgesic natural products from the sea whip Pseudopterogorgia elisabethae. J. Org. Chem. 1986, 51, 5140–5145. [Google Scholar]
- Newton, C.G.; Sherburn, M.S. Total synthesis of the pseudopterosin aglycones. Nat. Prod. Rep. 2015, 32, 865–876. [Google Scholar]
- Zhang, X.; Fang, X.; Xu, M.; Lei, Y.; Wu, Z.; Hu, X. Enantioselective total synthesis of pseudopteroxazole and ileabethoxazole. Angew. Chem. Int. Ed. 2019, 58, 7845–7849. [Google Scholar] [CrossRef]
- Rodríguez, A.D.; Ramírez, C.; Rodríguez, I.I.; Barnes, C.L. Novel terpenoids from the West Indian sea whip Pseudopterogorgia elisabethae (Bayer). Elisapterosins A and B: Rearranged diterpenes possessing an unprecedented cagelike framework. J. Org. Chem. 2000, 65, 1390–1398. [Google Scholar] [CrossRef]
- Waizumi, N.; Stankovic, A.R.; Rawal, V.H. A general strategy to elisabethane diterpenes: Stereocontrolled synthesis of elisapterosin B via oxidative cyclization of an elisabethin precursor. J. Am. Chem. Soc. 2003, 125, 13022–13023. [Google Scholar]
- Lazerwith, S.E.; Johnson, T.W.; Corey, E.J. Syntheses and stereochemical revision of pseudopterosin G–J aglycon and helioporin E. Org. Lett. 2000, 2, 2389–2392. [Google Scholar] [CrossRef]
- Rodríguez, I.I.; Shi, Y.-P.; García, O.J.; Rodríguez, A.D.; Mayer, A.M.S.; Sánchez, J.A.; Ortega-Barria, E.; González, J. New pseudopterosin and seco-pseudopterosin diterpene glycosides from two Colombian isolates of Pseudopterogorgia elisabethae and their diverse biological activities. J. Nat. Prod. 2004, 67, 1672–1680. [Google Scholar] [CrossRef]
- Salae, A.-W.; Boonnak, N. Obtusinones D and E, linear and angular fused dimeric icetexane diterpenoids from Premna obtusifolia roots. Tetrahedron Lett. 2013, 54, 1356–1359. [Google Scholar]
- Fu, G.-M.; Qin, H.-L.; Yu, S.-S.; Yu, B.-Y. Yuexiandajisu D, a novel 18-nor-rosane-type dimeric diterpenoid from Euphorbia ebracteolata Hayata. J. Asian Nat. Prod. Res. 2006, 8, 29–34. [Google Scholar] [CrossRef]
- Gao, C.; Han, L.; Zheng, D.; Jin, H.; Gai, C.; Wang, J.; Zhang, H.; Zhang, L.; Fu, H. Dimeric abietane diterpenoids and sesquiterpenoid lactones from Teucrium viscidum. J. Nat. Prod. 2015, 78, 630–638. [Google Scholar]
- Galli, B.; Gasparrini, F.; Lanzotti, V.; Misiti, D.; Riccio, R.; Villani, C.; Guan-fu, H.; Zhong-wu, M.; Wan-fen, Y. Grandione, a new heptacyclic dimeric diterpene from Torreya grandis Fort. Tetrahedron 1999, 55, 11385–11394. [Google Scholar]
- Nair, V.; Menon, R.S.; Biju, A.T.; Abhilash, K.G. 1,2-Benzoquinones in Diels–Alder reactions, dipolar cycloadditions, nucleophilic additions, multicomponent reactions and more. Chem. Soc. Rev. 2012, 41, 1050–1059. [Google Scholar] [CrossRef]
- Rodríguez, A.D.; Ramírez, C.; Rodríguez, I.I. Elisabatins A and B: New amphilectane-type diterpenes from the West Indian sea whip Pseudopterogorgia elisabethae. J. Nat. Prod. 1999, 62, 997–999. [Google Scholar] [CrossRef]
- Baran, P.; Raptis, R.G.; Rodríguez, A.D.; Rodríguez, I.I.; Shi, Y.-P. Structural characterization of elisabatin B and elisabatin C. J. Chem. Crystallogr. 2003, 33, 711–718. [Google Scholar] [CrossRef]
- Walker, D.H. Small-molecule inhibitors of cyclin-dependent kinases: Molecular tools and potential therapeutics. Curr. Top. Microbiol. Immunol. 1998, 227, 149–165. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | δH, Mult, J in Hz | δC, Mult b | Atom | δH, Mult, J in Hz | δC, Mult b |
|---|---|---|---|---|---|
| 1 | 3.60, br dd, 9.3, 8.4 | 36.8, d | 1′ | 61.6, s | |
| 2α | 1.22, m | 40.2, t | 2′ | 141.1, s | |
| 2β | 1.94, m | 3′ | 3.14, m | 28.7, d | |
| 3 | 1.21, m | 34.0, d | 4α’ | 1.46, m | 24.3, t |
| 4 | 2.01, m | 44.6, d | 4β’ | 1.78, m | |
| 5α | 0.95, m | 27.7, t | 5α’ | 1.55, br dd, 11.4, 7.4 | 19.3, t |
| 5β | 2.03, m | 5β’ | 1.80, dd, 11.4, 5.7 | ||
| 6α | 2.15, ddd, 13.0, 9.1, 4.1 | 32.0, t | 6′ | 2.35, br dd, 10.4, 5.7 | 40.3, d |
| 6β | 1.30, ddd, 13.0, 9.1, 4.1 | 7′ | 2.05, m | 41.7, d | |
| 7 | 3.20, m | 28.6, d | 8α’ | 2.22, ddd, 12.4, 12.2, 6.1 | 44.5, t |
| 8 | 126.2, s | 8β’ | 0.97, m | ||
| 9 | 147.1, s | 9′ | —c | 53.4, d c | |
| 10 | 137.2, s | 10′ | —c | 60.7, d c | |
| 11 | 129.6, s c | 11′ | ~85.0, s c | ||
| 12 | 129.6, s c | 12′ | 1.01, br s | 26.8, q c | |
| 13 | 135.9, s | 13′ | 1.02, br s | 26.9, q c | |
| 14 | 4.94, br d, 9.3 | 131.4, d | 14′ | 203.1, s | |
| 15 | 128.3, s | 15′ | 69.2, s c | ||
| 16 | 1.66, br s | 25.4, q | 16′ | ~196.0, s c | |
| 17 | 1.70, br s | 17.5, q | 17′ | 146.9, s | |
| 18 | 1.02, br s | 20.0, q | 18′ | 1.16, d, 7.1 | 17.7, q |
| 19 | 1.25, d, 6.7 | 23.1, q | 19′ | 1.06, d, 6.4 | 18.1, q |
| 20 | 1.96, s | 15.6, q | 20′ | 1.66, s | 15.6, q |
| 9-OH | 5.97, s, exchangeable | 17′-OH | 6.18, br s, exchangeable |
| Atom | δH, Mult, J in Hz | δC, Mult b | Atom | δH, Mult, J in Hz | δC, Mult b |
|---|---|---|---|---|---|
| 1 | 3.65, dd, 8.8, 8.4 | 36.7, d | 1′ | 144.7, s | |
| 2α | 1.18, m | 40.3, t | 2α’ | 1.82, m | 42.2, t |
| 2β | 1.94, dd, 10.4, 1.9 | 2β’ | 2.12, m | ||
| 3 | 1.20, m | 34.2, d | 3′ | 1.36, m | 32.8, d |
| 4 | 1.98, m | 44.4, d | 4′ | 1.80, m | 44.2, d |
| 5α | 0.97, m | 28.3, t | 5α’ | 1.17, m | 25.3, t |
| 5β | 2.07, dd, 7.5, 3.7 | 5β’ | 2.00, m | ||
| 6α | 2.17, ddd, 12.6, 7.9, 4.3 | 31.8, t | 6α’ | 2.09, dd, 7.4, 3.7 | 31.0, t |
| 6β | 1.30, dd, 8.7, 4.3 | 6β’ | 1.12, m | ||
| 7 | 3.35, q, 7.7 | 27.6, d | 7′ | 2.82, q, 6.9 | 29.3, d |
| 8 | 126.3, s | 8′ | 129.3, s | ||
| 9 | 137.2, s | 9′ | 191.2, s | ||
| 10 | 137.8, s | 10′ | 90.1, s | ||
| 11 | 122.3, s | 11′ | 78.8, s | ||
| 12 | 129.7, s | 12′ | 132.5, s | ||
| 13 | 132.3, s | 13′ | 157.9, s | ||
| 14 | 5.00, dd, 10.2, 1.0 | 131.8, d | 14′ | 5.71, br s | 126.4, d |
| 15 | 127.6, s | 15′ | 129.4, s | ||
| 16 | 1.63, d, 1.0 | 25.3, q | 16′ | 1.76, d, 1.0 | 25.3, q |
| 17 | 1.68, d, 0.8 | 17.5, q | 17′ | 1.50, d, 0.8 | 19.6, q |
| 18 | 1.00, d, 5.9 | 20.0, q | 18′ | 0.95, d, 6.5 | 19.1, q |
| 19 | 1.21, d, 6.7 | 23.3, q | 19′ | 1.22, d, 6.6 | 22.0, q |
| 20 | 1.84, s | 11.5, q | 20′ | 1.45, s | 21.0, q |
| 10′-OH | 4.70, s, exchangeable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, I.I.; Rodríguez, A.D.; Barnes, C.L. Isolation, Structural Analysis and Biological Activity Assays of Biselisabethoxanes A and B: Two Dissymmetric Bis-Diterpenes from the Southwestern Caribbean Sea Gorgonian Coral Pseudopterogorgia elisabethae. Molecules 2022, 27, 7879. https://doi.org/10.3390/molecules27227879
Rodríguez II, Rodríguez AD, Barnes CL. Isolation, Structural Analysis and Biological Activity Assays of Biselisabethoxanes A and B: Two Dissymmetric Bis-Diterpenes from the Southwestern Caribbean Sea Gorgonian Coral Pseudopterogorgia elisabethae. Molecules. 2022; 27(22):7879. https://doi.org/10.3390/molecules27227879
Chicago/Turabian StyleRodríguez, Ileana I., Abimael D. Rodríguez, and Charles L. Barnes. 2022. "Isolation, Structural Analysis and Biological Activity Assays of Biselisabethoxanes A and B: Two Dissymmetric Bis-Diterpenes from the Southwestern Caribbean Sea Gorgonian Coral Pseudopterogorgia elisabethae" Molecules 27, no. 22: 7879. https://doi.org/10.3390/molecules27227879
APA StyleRodríguez, I. I., Rodríguez, A. D., & Barnes, C. L. (2022). Isolation, Structural Analysis and Biological Activity Assays of Biselisabethoxanes A and B: Two Dissymmetric Bis-Diterpenes from the Southwestern Caribbean Sea Gorgonian Coral Pseudopterogorgia elisabethae. Molecules, 27(22), 7879. https://doi.org/10.3390/molecules27227879