
In Vitro Screening for Anti-Acetylcholinesterase and Antioxidant Activities of Hottonia palustris L. Extracts and Their Unusual Flavonoids





Abstract
:1. Introduction
2. Results and Discussion
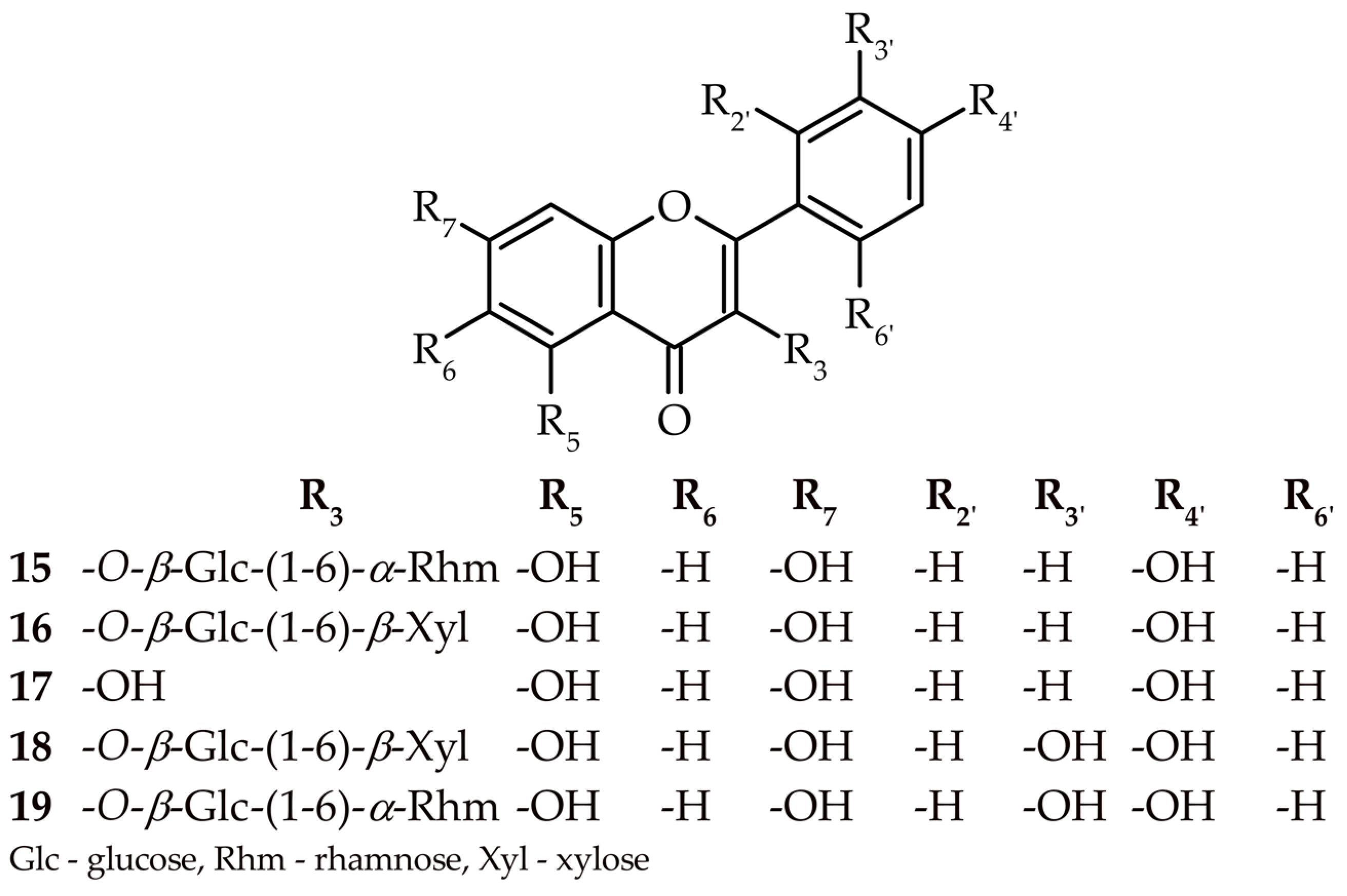
2.1. Determination of Isolated Compounds 8–19
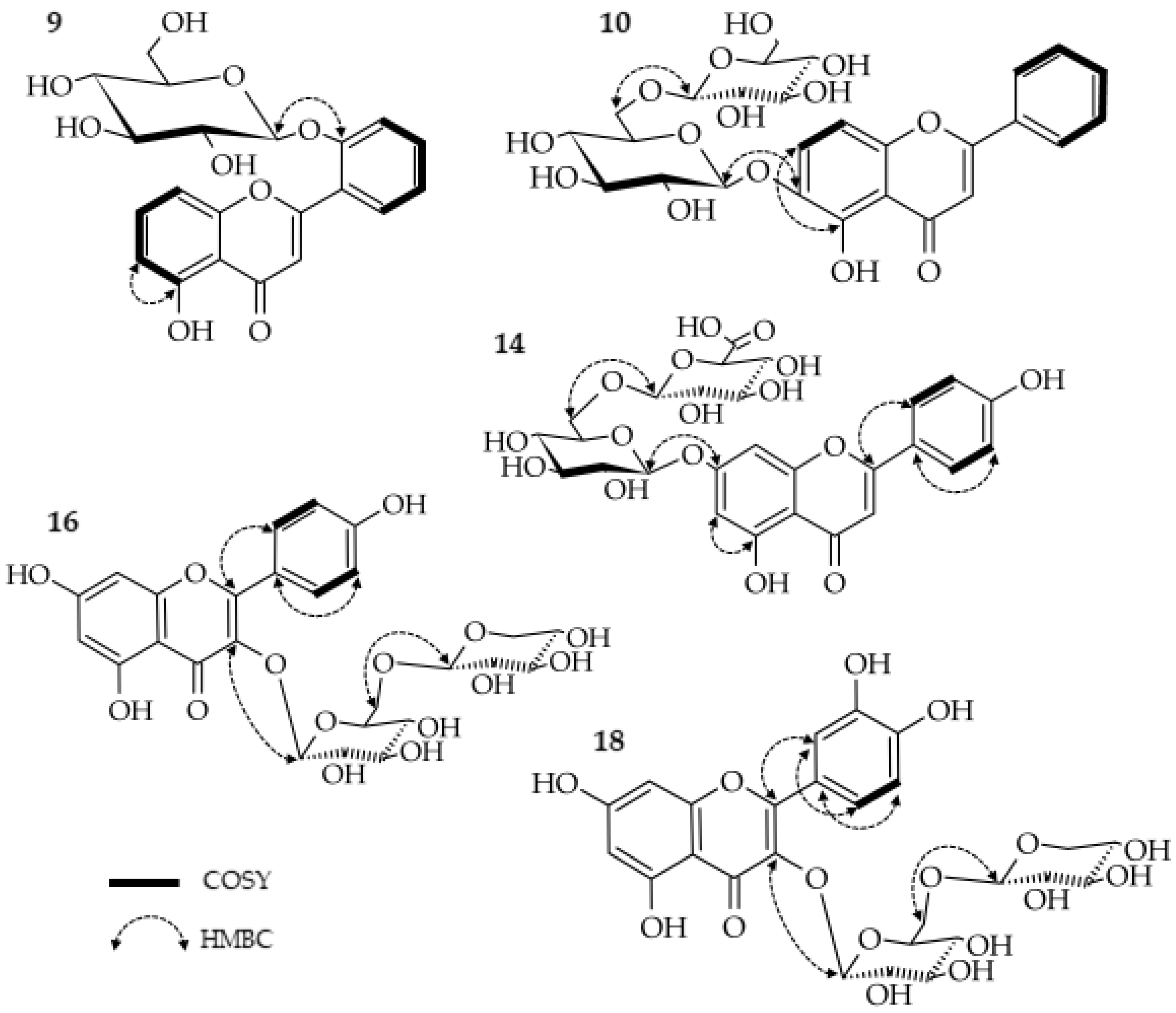
2.1.1. 5,2′-Dihydroxyflavone 2′-O-β-glucopyranoside (9)
2.1.2. 5,6-Dihydroxyflavone 6-O-(6″-O-β-glucopyranosyl)-β-glucopyranoside (hottonioside A) (10)
2.1.3. Apigenin 7-O-(2″-O-β-glucuronide)-β-glucopyranoside (14)
2.1.4. Kaempferol 3-O-(6″-O-β-xylopyranosyl)-β-glucopyranoside (16)
2.1.5. Quercetin 3-O-(6″-O-β-xylopyranosyl)-β-glucopyranoside (18)
2.2. Evaluation of H. palustris Metabolome
2.2.1. Qualitative LC-PDA-HRMS Analysis of H. palustris Extracts and Fractions
2.2.2. Quantitative Characteristics (TPC, TFC, TPAC, TTC) of H. palustris Extracts and Fractions
2.3. Assessment of the Antioxidant and Acetylcholinesterase Inhibiting Activities
3. Materials and Methods
3.1. Chemicals and General Experimental Procedures
3.2. Plant Materials
3.3. Preparation of Extracts and Isolation Procedure
3.3.1. Extraction
3.3.2. Isolation Using the MPLC and HPLC Techniques
3.3.3. Hydrolysis of the Glycosides
3.4. Elucidation of Chemical Structure of Compounds 8–19
3.5. Conditions of Chromatographic Analysis
3.5.1. Sample Preparation
3.5.2. LC-PDA-HRMS Data Acquisition
3.5.3. Preparative HPLC
3.6. Quantitative Characteristics of H. palustris Extracts
3.6.1. Determination of the Total Phenol Content (TPC)
3.6.2. Determination of the Total Flavonoid Content (TFC)
3.6.3. Determination of Total Phenolic Acid Content (TPAC)
3.6.4. Determination of the Total Tannin Content (TTC)
3.7. Antioxidant Activity (DPPH, ABTS, CUPRAC, FRAP)
3.7.1. Free Radical Scavenging Activity (DPPH Assay)
3.7.2. ABTS [2,2′-azino-bis(3-ethylbenzothazoline-6-sulphonic acid)] Radical Cation Scavenging Activity
3.7.3. Ferric-Reducing Antioxidant Power Method (FRAP)
3.7.4. Cupric Ion Reducing Method (CUPRAC)
3.8. Acetylcholinesterase Inhibitory Activity
4. Statistical Analysis and Simulated Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| UV | ultraviolet spectroscopy |
| HRMS | high-resolution mass spectrometry |
| NMR | nuclear magnetic resonance |
| 2D NMR | two-dimensional nuclear magnetic resonance |
| COSY | correlation spectroscopy |
| HSQC | heteronuclear single quantum coherence |
| HMBC | heteronuclear multiple bond correlation |
| DEPT | distortionless enhancement by polarization transfer |
| UV–Vis | ultraviolet–visible spectroscopy |
| TPC | total phenolic content |
| TFC | total flavonoid content |
| TPAC | total phenolic acid content |
| TTC | total tannin content |
| ABTS | 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) |
| FRAP | ferrous reducing antioxidant potential |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| CUPRAC | cupric reducing antioxidant capacity |
| FeCl3 | ferric (III) chloride |
| AChE | acetylcholinesterase |
| ACh iodide | acetylcholine iodide |
| SD | standard deviation |
| IC50 | concentration at which a substance exerts half of its maximal inhibitory effect |
References
- Elliott, S. A Sketch of the Botany of South Carolina and Georgia; J.R. Schenck: Charleston, SC, USA, 1821. [Google Scholar]
- World Flora Online. Hottonia inflata Elliott. Available online: http://www.worldfloraonline.org/taxon/wfo-0001105360 (accessed on 16 September 2022).
- Ramesh, S.; Rajan, R.; Santhanam, R. Freshwater Phytopharmaceutical Compounds; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- World Flora Online. Hottonia palustris L. Available online: http://www.worldfloraonline.org/taxon/wfo-0000725040 (accessed on 16 September 2022).
- von Linneé, C.; Salvius, L. Species Plantarum: Exhibentes Plantas Rite Cognitas, Ad Genera Relatas, Cum Differentiis Specificis, Nominibus Trivialibus, Synonymis Selectis, Locis Natalibus, Secundum Systema Sexuale Sigestas; Impensis GC Nauk: Stockholm, Sweden, 1797; Volume 2. [Google Scholar]
- Lauber, K.; Wagner, G.; Gygax, A. Flora Helvetica—Illustrierte Flora der Schweiz, 6th ed.; Haupt Verlag: Bern, Switzerland, 2018. [Google Scholar]
- Strawa, J.W.; Jakimiuk, K.; Szoka, Ł.; Brzezinski, K.; Drozdzal, P.; Pałka, J.A.; Tomczyk, M. New polymethoxyflavones from Hottonia palustris evoke DNA biosynthesis-inhibitory activity in an oral squamous carcinoma (SCC-25) cell line. Molecules 2022, 27, 4415. [Google Scholar] [CrossRef] [PubMed]
- Broda, B.; Mowszowicz, J. Przewodnik Do Oznaczania Roślin Leczniczych, Trujących I Użytkowych; Wydawnictwo Lekarskie PZWL: Warszawa, Poland, 1996. [Google Scholar]
- World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- World Health Organization. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Li, R.S.; Wang, X.B.; Hu, X.J.; Kong, L.Y. Design, Synthesis and evaluation of flavonoid derivatives as potential multifunctional acetylcholinesterase inhibitors against Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2013, 23, 2636–2641. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Hunter, J.M. Anticholinesterases and anticholinergic drugs. Contin. Educ. Anaesth. Crit. Care Pain 2004, 4, 164–168. [Google Scholar] [CrossRef] [Green Version]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Ding, H.-Y.; Chen, Y.-Y.; Chang, W.-L.; Lin, H.-C. Flavonoids from the flowers of Pueraria lobata. J. Chin. Chem. Soc. 2004, 51, 1425–1428. [Google Scholar] [CrossRef]
- Mainka, M.; Czerwińska, M.E.; Osińska, E.; Ziaja, M.; Bazylko, A. Screening of antioxidative properties and inhibition of inflammation-linked enzymes by aqueous and ethanolic extracts of plants traditionally used in wound healing in Poland. Antioxidants 2021, 10, 698. [Google Scholar] [CrossRef]
- Alinezhad, H.; Zare, M.; Nabavi, S.F.; Naqinezhad, A.; Nabavi, S.M. Antioxidant, antihemolytic, and inhibitory activities of endemic Primula heterochroma against Fe2+ - induced lipid peroxidation and oxidative stress in rat brain in vitro. Pharm. Biol. 2012, 50, 1391–1396. [Google Scholar] [CrossRef]
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones and flavonols antiradical structure—Activity relationship—A quantum chemical study. Antioxidants 2020, 9, 461. [Google Scholar] [CrossRef]
- Burda, S.; Oleszek, W. Antioxidant and antiradical activities of flavonoids. J. Agric. Food Chem. 2001, 49, 2774–2779. [Google Scholar] [CrossRef]
- dos Santos, T.C.; Gomes, T.M.; Pinto, B.A.S.; Camara, A.L.; de Andrade Paes, A.M. Naturally occurring acetylcholinesterase inhibitors and their potential use for Alzheimer’s disease. Front. Pharmacol. 2018, 9, 1192. [Google Scholar] [CrossRef]
- Uriarte-Pueyo, I.; Calvo, M. Flavonoids as acetylcholinesterase inhibitors. Curr. Med. Chem. 2011, 18, 5289–5302. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Hay, A.-E.; Marston, A.; Hostettmann, K. Acetylcholinesterase-inhibitory activity of linarin from Buddleja davidii, structure-activity relationships of related flavonoids and chemical investigation of Buddleja nitida. Pharm. Biol. 2008, 46, 596–601. [Google Scholar] [CrossRef]
- Strawa, J.; Wajs-Bonikowska, A.; Jakimiuk, K.; Waluk, M.; Poslednik, M.; Nazaruk, J.; Tomczyk, M. Phytochemical examination of woolly burdock Arctium tomentosum leaves and flower heads. Chem. Nat. Compd. 2020, 56, 345–347. [Google Scholar] [CrossRef]
- Strawa, J.W.; Jakimiuk, K.; Tomczyk, M. Zapotin, a polymethoxyflavone, with potential therapeutic attributes. Int. J. Mol. Sci. 2021, 22, 13227. [Google Scholar] [CrossRef]
- Abraham, M.H.; Acree, W.E.; Earp, C.E.; Vladimirova, A.; Whaley, W.L. Studies on the hydrogen bond acidity and other descriptors and properties for hydroxyflavones and hydroxyisoflavones. J. Mol. Liq. 2015, 208, 363–372. [Google Scholar] [CrossRef]
- Wollenweber, E.; Mann, K.; Inuma, M.; Tanaka, T.; Mizuno, M. 8,2′-Dihydroxyflavone from Primula pulverulenta. Phytochemistry 1988, 27, 1483–1486. [Google Scholar] [CrossRef]
- Harborne, J.B. The Flavonoids Advances in Research since 1986, 1st ed.; Routledge: New York, NY, USA, 2017. [Google Scholar]
- Agrawal, P.K. Carbon-13 NMR of Flavonoids, 1st ed.; Elseiver: New York, NY, USA, 1989. [Google Scholar]
- Ricelli, A.; De Angelis, M.; Primitivo, L.; Righi, G.; Sappino, C.; Antonioletti, R. Role of some food-grade synthesized flavonoids on the control of ochratoxin A in Aspergillus carbonarius. Molecules 2019, 24, 2553. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Moon, B.-H.; Lee, E.; Lee, Y.; Yoon, Y.; Ahn, J.-H.; Lim, Y. 1H and 13C-NMR data of hydroxyflavone derivatives. Magn. Reson. Chem. 2007, 45, 674–679. [Google Scholar] [CrossRef]
- Ganbaatar, C.; Gruner, M.; Mishig, D.; Duger, R.; Schmidt, A.W.; Knölker, H.-J. Flavonoid glycosides from the aerial parts of Polygonatum odoratum (Mill.) Druce growing in Mongolia. Open Nat. Prod. J. 2015, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ciganović, P.; Jakimiuk, K.; Tomczyk, M.; Končić, M.Z. Glycerolic licorice extracts as active cosmeceutical ingredients: Extraction optimization, chemical characterization, and biological activity. Antioxidants 2019, 8, 445. [Google Scholar] [CrossRef]
- Jakimiuk, K.; Strawa, J.W.; Granica, S.; Locatelli, M.; Tartaglia, A.; Tomczyk, M. Determination of flavonoids in selected Scleranthus species and their anti-collagenase and antioxidant potential. Molecules 2022, 27, 2015. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, D.M.; Uysal, S.; Aktumsek, A.; Granica, S.; Zengin, G.; Ceylan, R.; Locatelli, M.; Tomczyk, M. In vitro enzyme inhibitory properties, antioxidant activities and phytochemical profile of Potentilla thuringiaca. Phytochem. Lett. 2017, 20, 365–372. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C No. | 9 | 10 | 14 | 16 | 18 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δ C | δ H | δ C | δ H | δ C | δ H | δ C | δ H | δ C | δ H | |
| 2 | 161.85 C | - | 164.58 C | - | 164.26 C | - | 157 C | - | 156.19 C | - |
| 3 | 110.78 B | 7.16, s | 104.97 B | 7.03, s | 103.05 B | 6.85, s | 133.69 C | - | 133.26 C | - |
| 4 | 183.29 C | - | 183.85 C | - | 182.06 C | - | 177.86 C | - | 177.23 C | - |
| 5 | 159.8 C | - | 148.51 C | - | 161.13 C | - | 161.64 C | - | 161.14 C | - |
| 6 | 110.69 B | 6.82, d, J = 8.16 | 140.59 C | - | 99.54 B | 6.45, s | 99.19 B | 6.19, s | 98.88 B | 6.15, s |
| 7 | 135.9 B | 7.68, t, J = 8.28, 8.16 | 107.11 B | 7.72, d, J = 9.03 | 162.76 C | - | 164.69 C | - | 164.94 C | - |
| 8 | 107.53 B | 7.38, d, J = 8.28 | 124.14 B | 7.18, d, J = 9.03 | 95.25 B | 6.82, s | 94.21 B | 6.41, s | 93.7 B | 6.35, s |
| 9 | 156.2 C | - | 150.49 C | - | 156.83 C | - | 156.93 C | - | 156.41 C | - |
| 10 | 110.03 C | - | 110.61 C | - | 105.4 C | - | 104.49 C | - | 103.72 C | - |
| 5-OH | 12.71, s | - | 12.73, s | - | 12.96, s | - | 12.59, s | - | 12.61, s | |
| 4′-OH | - | - | - | - | 10.44, s | - | 10.24, s | - | nd | |
| 1′ | 120.07 C | - | 130.84 C | - | 121.04 C | - | 121.35 C | - | 121.08 C | - |
| 2′ | 155.63 | - | 126.84 B | 8.09, d, J = 7.03 | 128.61 B | 7.95, d, J = 8.53 | 131.42 B | 8.03, d, J = 8.66 | 116.15 B | 7.56, s |
| 3′ | 115.59 B | 7.17, d, J = 6.78 A | 129.46 B | 7.6, m | 115.99 B | 6.94, d, J = 8.53 | 115.57 B | 6.88, d, J = 8.66 | 144.82 C | - |
| 4′ | 133.23 B | 7.58, t, J = 7.4, 7.4 | 132.58 B | 7.6, m | 161.36 C | - | 160.4 C | - | 148.6 C | - |
| 5′ | 122.04 B | 7.23, t, J = 7.4, 7.4 | 129.46 B | 7.6, m | 115.99 B | 6.94, d, J = 8.53 | 115.57 B | 6.88, d, J = 8.66 | 115.24 B | 6.83, d, J = 8.03 |
| 6′ | 129.26 B | 7.94, d, J = 6.78 | 126.84 B | 8.09, d, J = 7.03 | 128.61 B | 7.95, d, J = 8.53 | 131.42 B | 8.03, d, J = 8.66 | 121.66 B | 7.58, d, J = 2.26 |
| 2′-O-glucosyl | 6-O-glucosyl | 7-O-glucosyl | 3-O-glucosyl | 3-O-glucosyl | ||||||
| 1″ | 100.27 | 5.13, d, J = 7.03 B | 101 | 4.92, d, J = 6.53 B | 98.29 | 5.2, d, J = 7.03 B | 101.48 | 5.37, d, J = 7.53 B | 100.94 | 5.37, d, J = 7.28 B |
| 2″ | 73.33 | 3.31 A,B | 73.81 | 2.96 A,B | 82.74 | 3.54 A,B | 73.61 | 2.85 A,B | 73.98 | 3.21 A,B |
| 3″ | 77.21 | 3.46 A,B | 76.96 | 3.05 A,B | 75.76 | 3.15 A,B | 76.73 | 3.22 A,B | 76.35 A | 3.24 A,B |
| 4″ | 69.56 | 3.2 A,B | 70.31 | 3.12 A,B | 69.14 | 3.26 A,B | 70.13 | 3.13 A,B | 69.76 | 2.98 A,B |
| 5″ | 76.72 | 3.32 A,B | 76.80 | 3.33 A,B | 76.98 | 3.5 A,B | 76.64 | 2.85 A,B | 76.21 | 2.87 A,B |
| 6″ | 60.59 | 3.72, d, J = 11.54 | 68.76 | 3.99, d, J = 9.79 | 60.48 | 3.73, d, J = 9.79 | 68.35 | 3.79, d, J = 11.29 | 69.41 | 3.81, d, J = 10.79 |
| 3.49, d, J = 5.27 B | 3.62 A,B | 3.57 A,B | 3.45 A,B | 3.44 A,B | ||||||
| - | 6″-O-glucosyl | glucuronide moiety | 2″-O-xylosyl | 2″-O-xylosyl | ||||||
| 1″′ | - | - | 103.5 B | 4.2, d, J = 7.78 | 104.44 B | 4.55, d, J = 7.78 | 104.13 B | 4.0, d, J = 7.28 | 103.65 B | 4.01, d, J = 7.28 |
| 2″′ | - | - | 73.43 | 3.29 A,B | 74.25 | 3.04 A,B | 74.55 | 3.16 A,B | 73.13 | 2.82 A,B |
| 3″′ | - | - | 76.82 | 3 A,B | 75.55 | 3.71 A,B | 76.7 A,D | 3.22 A,B | 76.35 A | 3.24 A,B |
| 4″′ | - | - | 70.02 | 3.30 A,B | 71.69 | 3.24 A,B | 69.86 | 2.81 A,B | 69.41 | 3.27 A,B |
| 5″′ | - | - | 76.16 | 3.61 A,B | 75.62 | 3.49 A,B | 65.79 | 3.51 A,B | 65.32 | 3.54 A,B |
| 2.76 A,B | 2.75 A,B | |||||||||
| 6″′ | - | - | 61.24 | 3.67 A,B | 170.67 | - | - | - | - | - |
| 3.41 A,B | ||||||||||
| No | Rt [min] | λ Max [nm] | Observed # | Formula | Δ [ppm] | Fragmentation | Compounds | Presence in HP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Negative/Positive | HP1 | HP2 | HP3 | HP4 | HP5 | HP6 | HP7 | HP8 | |||||||
| P1 | 17.35 | 256, 266, 348 | 623.1267 A | C27H28O17 | 2.26 | 623, 447, 285/625, 287 | kaempferol hex-uro derivative | x | x | x | x | ||||
| P2 | 19.87 | 268, 336 | 607.13 A | C27H28O16 | −0.49 | 607, 269/609, 271 | apigenin 7-O-(2″-O-β-gluc)-β-glu (14) | x | x | x | x | x | x | ||
| P3 | 22.27 | 254, 266, 295, 354 | 595.1317 A | C26H28O16 | 2.29 | 595, 300/597, 465, 303 | quercetin pent-hex | x | x | x | x | ||||
| P4 | 22.69 | 254, 266, 295, 354 | 595.1316 A | C26H28O16 | 2.29 | 595, 300/597, 465, 303 | quercetin 3-O-(6″-O-β-xyl)-β-glu (18) | x | x | x | x | x | |||
| P5 | 26.10 | 256, 266, 295, 354 | 609.146 A | C27H30O16 | 0.02 | 609, 300/611, 465, 303 | rutin (19) | x | x | x | x | x | x | ||
| P6 | 28.83 | 256, 264, 292, 352 | 579.1355 A | C26H28O15 | 3.6 | 579, 284/581, 417, 287 | kaempferol pent-hex | x | x | x | x | x | |||
| P7 | 29.56 | 256, 264, 292, 352 | 579.1356 A | C26H28O15 | 0.29 | 579, 284/581, 417, 287 | kaempferol 3-O-(6″-O-β-xyl)-β-glu (16) | x | x | x | x | x | |||
| P8 | 35.11 | 266, 294, 348 | 593.1514 A | C27H30O15 | 0.39 | 593, 284/595, 449, 287 | nicotiflorin (15) | x | x | x | x | x | x | ||
| P9 | 44.11 | 278, 314sh, 352sh | 577.1561 A | C27H30O14 | 0.11 | 577, 253/579, 417, 255 | hottonioside A (10) | x | x | x | |||||
| P10 | 46.63 | 278, 314sh, 352sh | 577.1565 A | C27H30O14 | −0.76 | 577, 254/579, 255 | dihydroxyflavone hex-hex | x | x | ||||||
| P11 | 48.65 | 257, 368 | 301.0354 A | C15H10O7 | 3.31 | 301/303 | quercetinS | x | x | x | |||||
| P12 | 48.87 | 268, 324 | 417.1187 B | C21H20O9 | 1.81 | -/417, 255 | 2′,5-dihydroxyflavone 2′-O-β-glu (9) | x | x | x | |||||
| P13 | 50.16 | 268, 290sh, 336 | 269.0457 A | C15H10O5 | 0.69 | 269/271 | apigeninS | x | x | x | x | x | x | ||
| P14 | 51.53 | 250, 265, 295sh, 320sh, 365 | 285.0407 A | C15H10O6 | 0.12 | 285/287 | kaempferol (17) | x | x | x | x | x | x | ||
| P15 | 53.46 | nd | 1249.6214 A | C60H98O27 | −0.19 | 1249, 328/- | sakurasosaponin | x | x | x | x | ||||
| P16 | 54.12 | nd | 1103.5664 A | C54H88O23 | 1.53 | 1103, 328/- | primulasaponin 1 | x | x | x | x | ||||
| P17 | 54.36 | nd | 1219.612 A | C54H88O23 | 1.53 | 1219/- | triterpene saponin derivative | x | x | x | x | ||||
| P18 | 54.54 | 282, 318, 360 | 253.0506 A | C15H10O4 | 2.21 | 253/255 | 5,6-dihydroxyflavone (12) | x | x | x | |||||
| P19 | 54.69 | nd | 957.5111 A | C41H82O24 | −1.1 | 957/- | triterpene saponin derivative | x | x | ||||||
| P20 | 54.9 | 272, 302, 334 | 253.0511 A | C15H10O4 | 2.03 | 253/255 | chrysin (13) | x | x | x | |||||
| P21 | 57.06 | 230, 258, 335 | 344.1251 B | C19H18O6 | −1.01 | -/343, 328, 313, 299 | zapotin (8) | x | x | x | x | x | x | ||
| P22 | 59.29 | 258, 333 | 313.0718 A | C17H14O6 | 0.67 | 313, 298, 283/315 | 5,6′-dihydroxy-2′,3′-dimethoxyflavone (7) | x | x | x | x | x | x | ||
| P23 | 59.95 | 259, 327 | 283.0612 A | C16 H12 O5 | 0.82 | 283, 268/285 | 2’,5-dihydroxy-6-methoxyflavone (6) | x | x | x | x | x | x | ||
| P24 | 66.27 | 268, 340 | 255.0506 A | C15H10O4 | 1.25 | 253/255 | 2’,5-dihydroxyflavone (11) | x | x | x | x | x | |||
| P25 | 72.64 | 257, 331 | 327.0887 A | C18H16O6 | 1.36 | 327/329, 299 | 5-hydroxy-2’,3’,6’-trimethoxyflavone (5) | x | x | x | x | ||||
| P26 | 81.71 | 260, 327 | 297.0772 A | C17H14O5 | 1.35 | 297/299 | 5-hydroxy-2’,6’-dimethoxyflavone (4) | x | x | x | x | x | |||
| P27 | 81.71 | 271, 336 | 239.0740 B | C15H10O3 | 4.78 | 237/239 | 5-hydroxyflavone (2) | x | x | x | x | x | |||
| P28 | 84.87 | 260, 327 | 267.0663 A | C16H12O4 | 0.93 | 267/269, 254 | 5-hydroxy-2′-methoxyflavone (3) | x | x | x | x | x | |||
| P29 | 91.94 | 252, 345 | 223.0765 A | C15H12O2 | 3.2 | 223/225 | 1,3-diphenylpropane-1,3-dione (1) | x | x | x | x | x | |||
| P30 | 97.48 | 310, 340, 460 | 687.2346 A | C52H30O2 | 1.02 | 687, 365/- | unknown | x | |||||||
| P31 | 99.46 | 280, 325, 395, 410 | 591.2645 A | C34H40O9 | −3.65 | 591/631, 593 | unknown | x | x | x | x | x | |||
| Extracts | TPC | TFC | TPAC | TTC |
|---|---|---|---|---|
| (mg Peq/g extract) A | (mg Leq/g extract) B | (mg CAeq/g extract) C | (mg Peq/g extract) A | |
| HP1 | 64.58 ± 1.01 | 7.10 ± 0.41 | 1.02 ± 0.06 | 2.94 ± 0.12 |
| HP2 | 49.54 ± 1.75 | 2.58 ± 0.16 | 0.58 ± 0.01 | 2.00 ± 0.31 |
| HP3 | 12.44 ± 0.20 | 1.62 ± 0.07 | 0.51 ± 0.01 | 2.11 ± 0.46 |
| HP4 | 67.81 ± 2.02 | 3.23 ± 0.29 | 0.80 ± 0.04 | 4.07 ± 0.82 |
| HP5 | 51.60 ± 2.37 | 2.69 ± 0.05 | 0.64 ± 0.03 | 5.40 ± 0.86 |
| HP6 | 64.52 ± 1.87 | 9.23 ± 0.12 | 3.03 ± 0.14 | 3.24 ± 0.58 |
| HP7 | 62.12 ± 1.67 | 7.63 ± 0.36 | 3.48 ± 0.21 | 1.51 ± 0.28 |
| HP8 | 58.31 ± 1.92 | 3.88 ± 0.12 | 0.76 ± 0.01 | 3.33 ± 0.82 |
| Extracts | DPPH A | ABTS A | FRAP B | CUPRAC A | AChE C |
|---|---|---|---|---|---|
| (µM Teq) | (µM Teq) | (mM Fe2+/mL) | (µM Teq) | IC50 (µg/mL) | |
| HP1 | 409.98 ± 4.83 | 945.81 ± 4.03 | 2.63 ± 0.04 | 354.42 ± 2.01 | 274.15 ± 0.98 |
| HP2 | 299.23 ± 3.59 | 774.72 ± 2.50 | 2.50 ± 0.04 | 375.08 ± 1.32 | 378.25 ± 3.22 |
| HP3 | 384.66 ± 3.65 | 773.90 ± 3.69 | 1.55 ± 0.15 | 79.67 ± 0.32 | na |
| HP4 | 452.16 ± 4.37 | 1132.21 ± 2.50 | 3.68 ± 0.21 | 385.19 ± 1.52 | 219.77 ± 0.37 |
| HP5 | 298.18 ± 3.59 | 1001.30 ± 2.28 | 2.36 ± 0.17 | 399.26 ± 2.63 | na |
| HP6 | 384.66 ± 1.83 | 1075.37 ± 3.71 | 4.35 ± 0.45 | 219.03 ± 0.76 | 260.24 ± 1.60 |
| HP7 | 494.35 ± 4.83 | 1099.42 ± 3.25 | 4.69 ± 0.14 | 306.95 ± 2.75 | 282.46 ± 0.47 |
| HP8 | 376.23 ± 3.48 | 945.81 ± 3.03 | 2.16 ± 0.06 | 129.79 ± 1.32 | 290.26 ± 1.51 |
| Compounds | DPPH A | ABTS A | FRAP B | CUPRAC A | AChE C |
|---|---|---|---|---|---|
| (µM Teq) | (µM Teq) | (mM Fe2+/mL) | (µM Teq) | IC50 (µM) | |
| 1 | 39.81 ± 2.76 | 300.12 ± 4.97 | na | 3.18 ± 0.32 | 144.83 ± 1.30 |
| 2 | 41.71 ± 2.28 | 317.47 ± 3.91 | na | na | 314.38 ± 1.72 |
| 3 | 39.39 ± 1.59 | 330.59 ± 4.04 | na | na | 279.70 ± 0.45 |
| 4 | 28.63 ± 2.40 | 326.76 ± 6.64 | na | na | 325.42 ± 3.21 |
| 5 | 34.32 ± 0.97 | 280.02 ± 1.55 | na | na | 284.61 ± 0.80 |
| 6 | 227.55 ± 2.85 | 781.28 ± 4.38 | 15.81 ± 0.85 | 366.29 ± 1.24 | 391.78 ± 1.06 |
| 7 | 37.85 ± 1.90 | 277.56 ± 2.88 | na | na | 390.83 ± 0.66 |
| 8 | 167.89 ± 1.08 | 756.95 ± 5.10 | 0.05 ± 0.01 | na | 464.45 ± 1.66 |
| 9 | 170.56 ± 2.49 | 204.59 ± 2.26 | na | 20.33 ± 1.57 | 399.72 ± 0.64 |
| 10 | 204.31 ± 3.31 | 624.94 ± 5.70 | na | 19.79 ± 1.03 | 376.11 ± 1.87 |
| 11 | 175.83 ± 2.83 | 661.30 ± 3.81 | 0.07 ± 0.01 | 44.50 ± 1.35 | 371.51 ± 0.66 |
| 12 | 1522.38 ± 7.31 | 1052.41 ± 5.51 | 54.25 ± 1.03 | 1060.42 ± 5.33 | 362.19 ± 0.39 |
| 13 | 227.51 ± 1.83 | 728.26 ± 1.08 | 0.08 ± 0.01 | na | 330.15 ± 1.77 |
| 14 | 341.42 ± 3.16 | 746.84 ± 1.85 | 1.63 ± 0.03 | 66.48 ± 0.56 | 417.78 ± 1.11 |
| 15 | 250.72 ± 1.65 | 702.29 ± 2.47 | 0.40 ± 0.06 | 56.81 ± 1.52 | 407.44 ± 1.41 |
| 16 | 300.29 ± 1.87 | 717.32 ± 3.47 | 0.67 ± 0.06 | 74.40 ± 1.49 | 364.34 ± 1.79 |
| 17 | 792.53 ± 7.31 | 1095.04 ± 1.85 | 36.87 ± 1.20 | 848.64 ± 3.49 | 276.90 ± 1.41 |
| 18 | 1336.75 ± 6.33 | 1147.52 ± 7.06 | 63.98 ± 1.75 | 1401.11 ± 2.64 | 302.85 ± 3.33 |
| 19 | 1467.53 ± 5.48 | 1149.16 ± 5.25 | 52.98 ± 1.35 | 1258.68 ± 3.49 | 445.32 ± 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strawa, J.W.; Jakimiuk, K.; Kita, Z.; Tomczyk, M. In Vitro Screening for Anti-Acetylcholinesterase and Antioxidant Activities of Hottonia palustris L. Extracts and Their Unusual Flavonoids. Molecules 2022, 27, 8034. https://doi.org/10.3390/molecules27228034
Strawa JW, Jakimiuk K, Kita Z, Tomczyk M. In Vitro Screening for Anti-Acetylcholinesterase and Antioxidant Activities of Hottonia palustris L. Extracts and Their Unusual Flavonoids. Molecules. 2022; 27(22):8034. https://doi.org/10.3390/molecules27228034
Chicago/Turabian StyleStrawa, Jakub W., Katarzyna Jakimiuk, Zuzanna Kita, and Michał Tomczyk. 2022. "In Vitro Screening for Anti-Acetylcholinesterase and Antioxidant Activities of Hottonia palustris L. Extracts and Their Unusual Flavonoids" Molecules 27, no. 22: 8034. https://doi.org/10.3390/molecules27228034