
Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted


 , , and
, , and
Abstract
:
1. Introduction
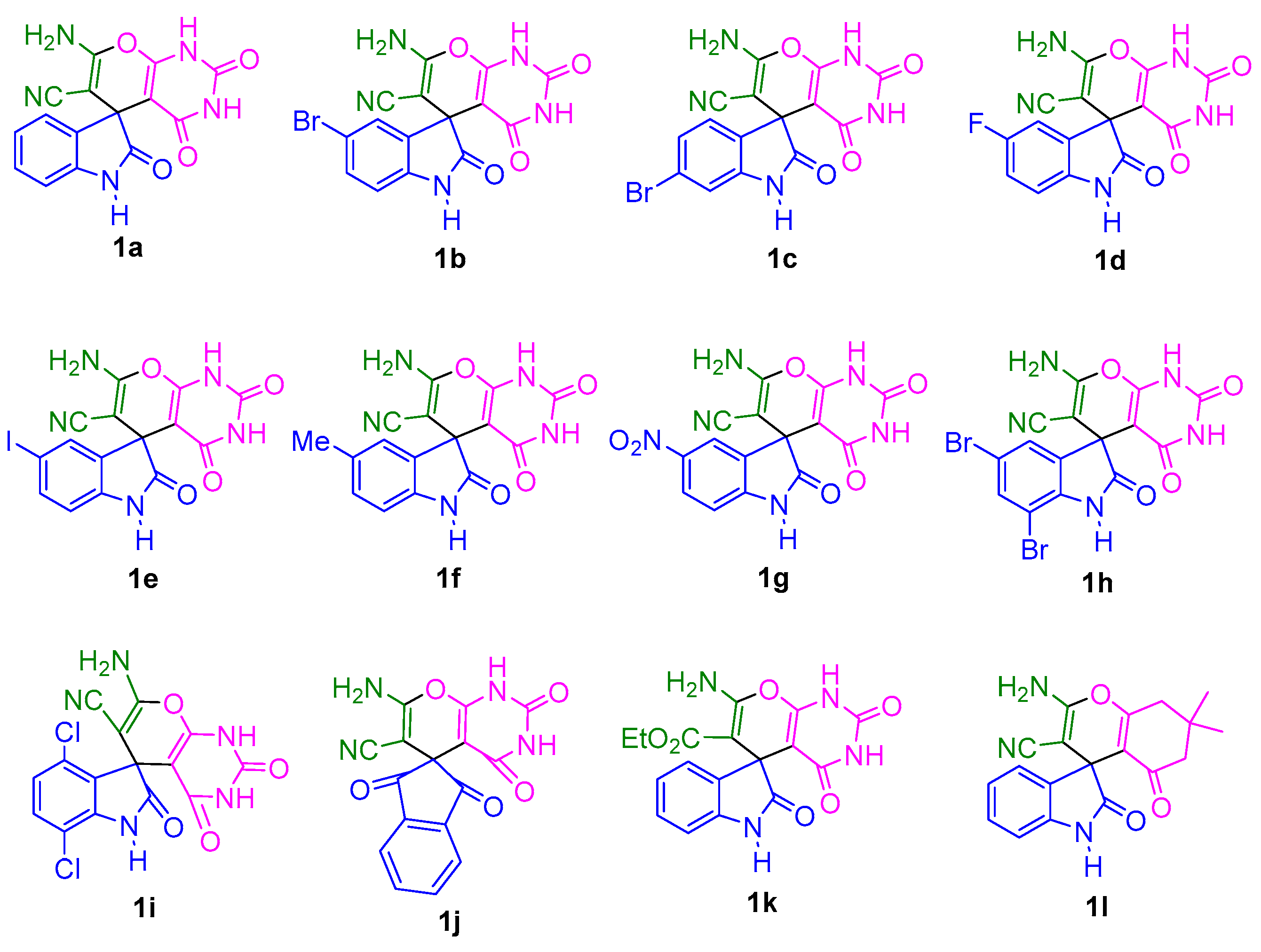
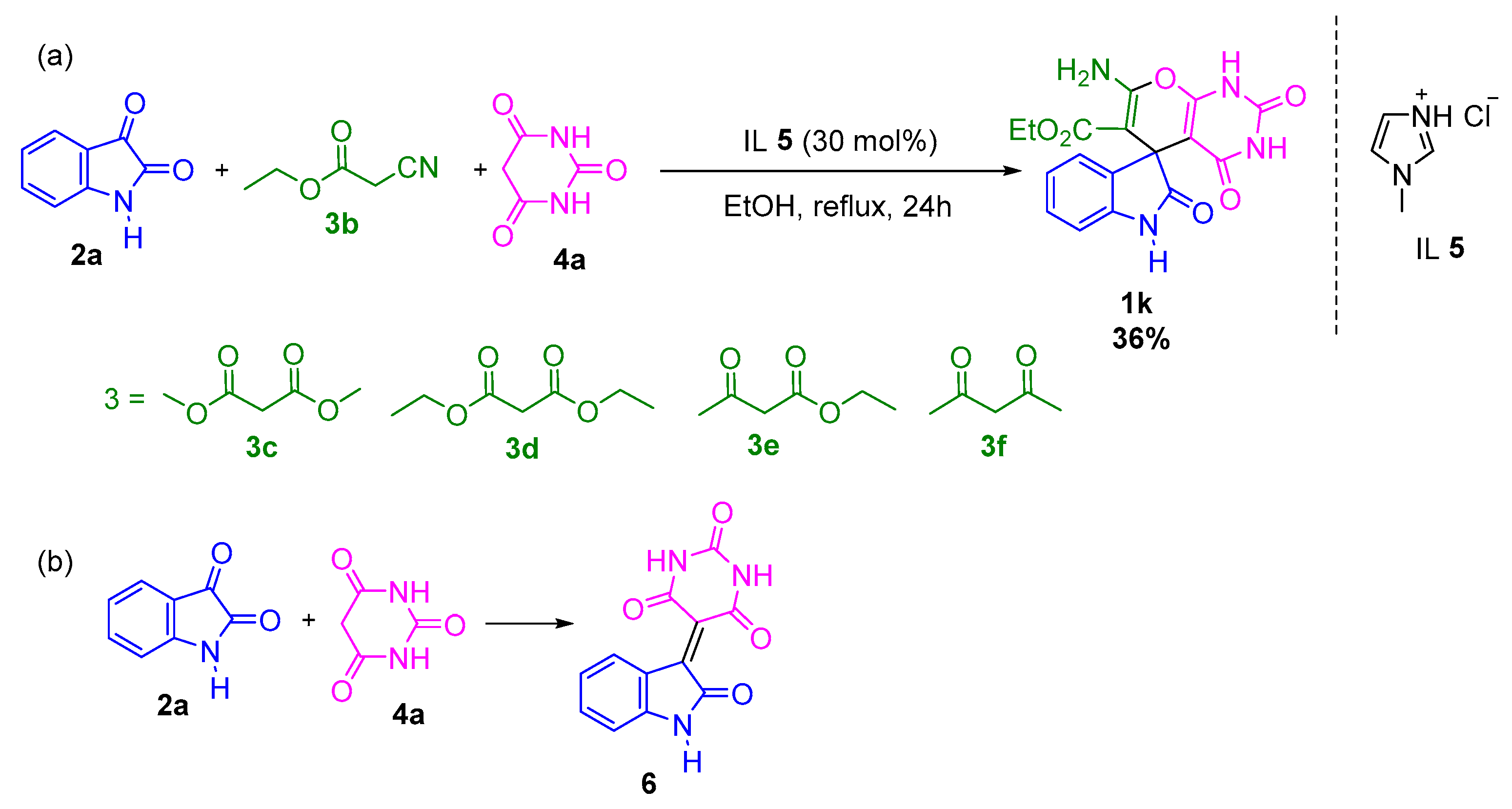
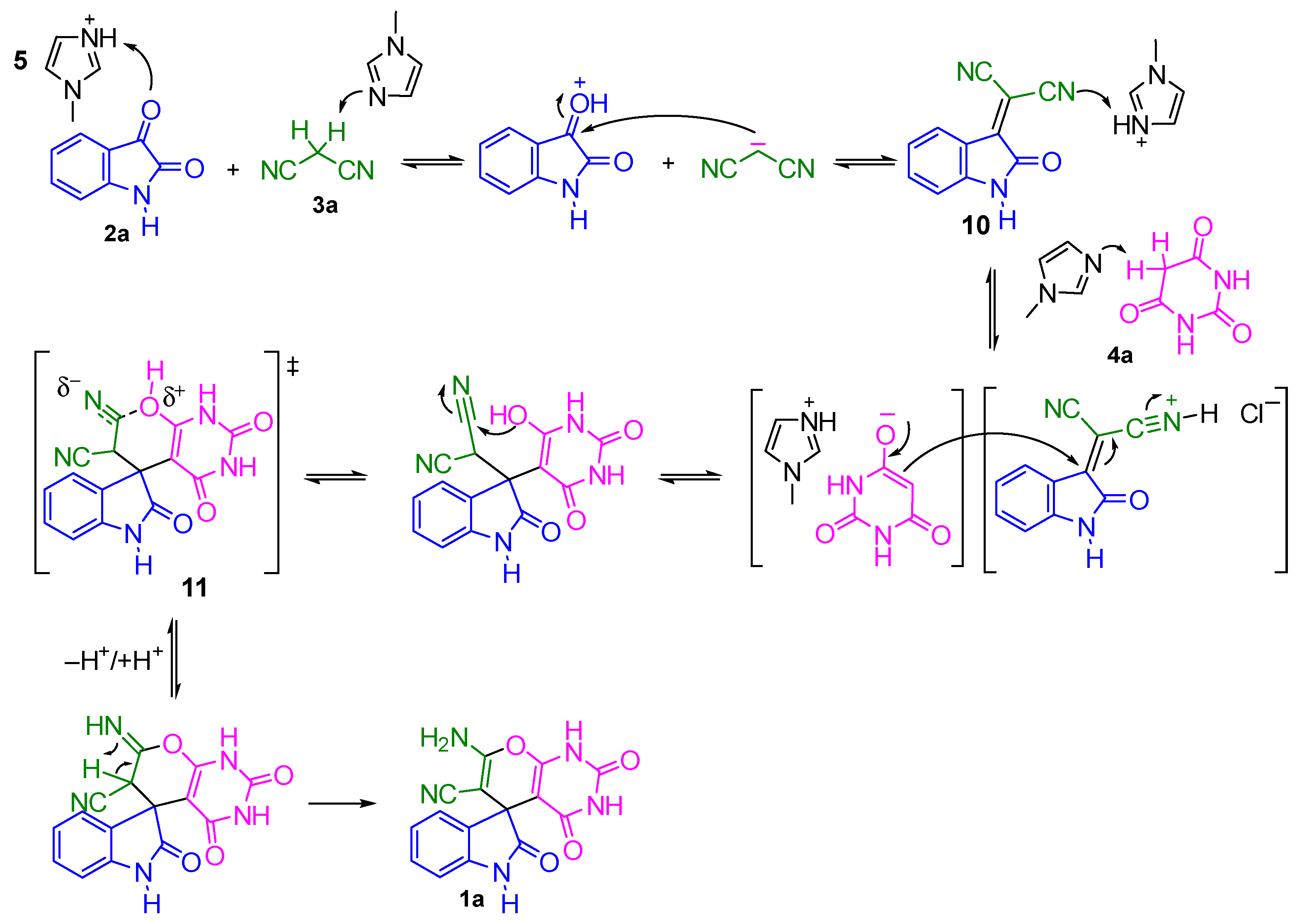
2. Results and Discussion
3. Experimental Session
3.1. Materials and Methods
3.2. General Procedure
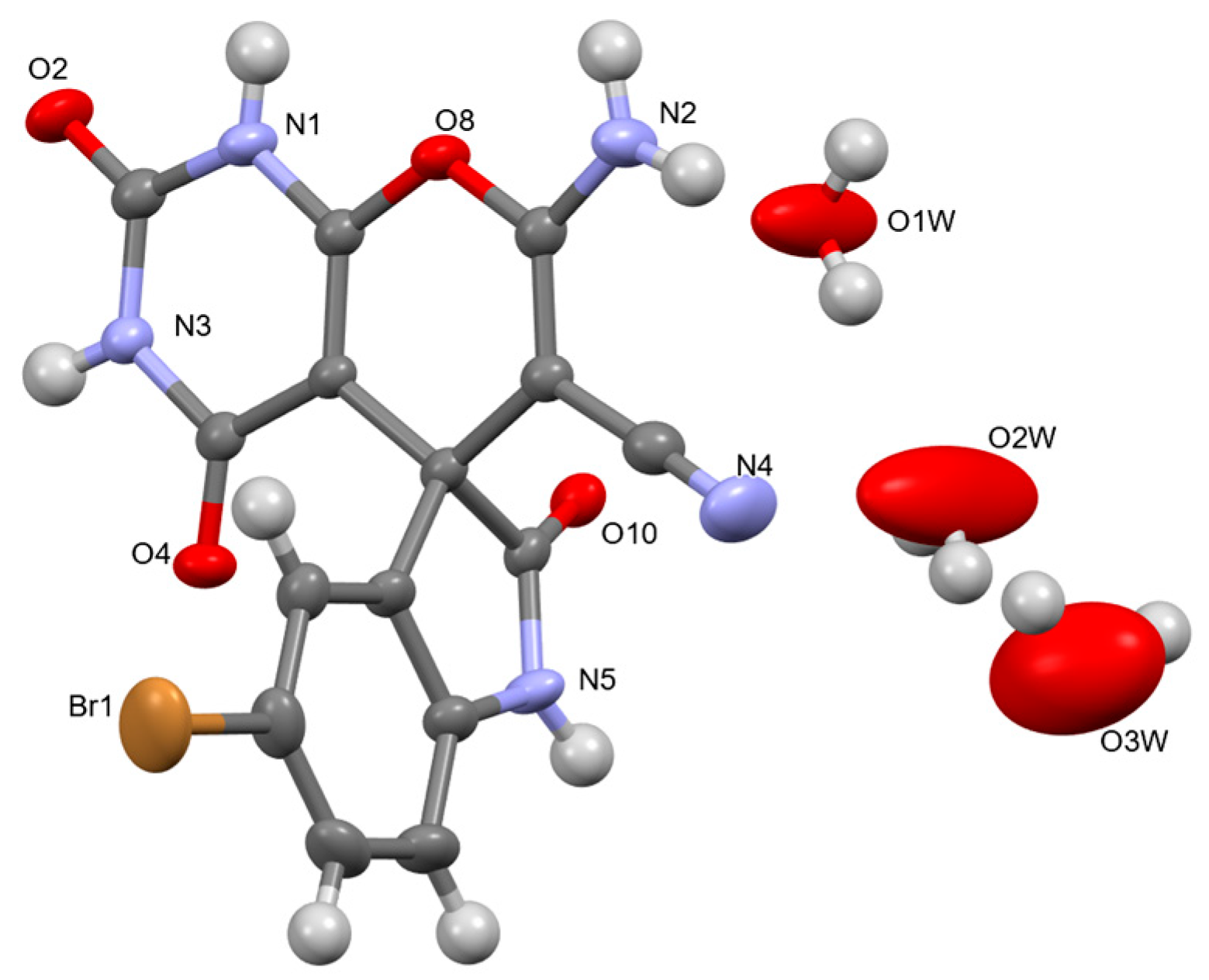
3.3. X-ray Diffraction Analysis
3.4. In Vitro Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pfau, A.S.; Plattner, P.A. Etudes sur les matières végétales volatiles X. Sur les vétivones, constituants odorants des essences de vétiver. Helv. Chim. Acta 1939, 22, 640–654. [Google Scholar] [CrossRef]
- Chupakhin, E.; Babich, O.; Prosekov, A.; Asyakina, L.; Krasavin, M. Spirocyclic Motifs in Natural Products. Molecules 2019, 24, 4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.K.; Baxendale, I.R. Total syntheses of natural products containing spirocarbocycles. Org. Biomol. Chem. 2015, 13, 9907–9933. [Google Scholar] [CrossRef] [Green Version]
- Raju, B.R.; Saikia, A.K. Asymmetric Synthesis of Naturally Occuring Spiroketals. Molecules 2008, 13, 1942–2038. [Google Scholar] [CrossRef]
- Quintavalla, A. Spirolactones: Recent Advances in Natural Products, Bioactive Compounds and Synthetic Strategies. Curr. Med. Chem. 2018, 25, 917–962. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphal, R.; Filho, E.V.; Medici, F.; Benaglia, M.; Greco, S.J. Stereoselective Domino Reactions in the Synthesis of Spiro Compounds. Synthesis 2022, 54, 2927–2975. [Google Scholar]
- Han, Y.-F.; Xia, M. Multicomponent Synthesis of Cyclic Frameworks on Knoevenagel-Initiated Domino Reactions. Curr. Org. Chem. 2010, 14, 379–413. [Google Scholar] [CrossRef]
- Dai, Y.; Di, J.; Hao, Z.; Meng, X.; Zhang, L. Synthesis of Spiro[benzo[b]thiophene-2(3H),1’-cyclopropan]-3-ones via Domino Reaction Between Thioaurones and Sulfur Ylides. Asian J. Org. Chem. 2021, 10, 1449–1453. [Google Scholar] [CrossRef]
- Zhou, H.-J.; Zhou, W.; Liu, X.-L.; Tian, Y.-P.; Wang, J.-X.; Zhou, Y. Catalytic Asymmetric Domino Michael/Annulation Reaction of Bifunctional Chromone Synthons with β,γ-Unsaturated α-Keto Esters: Rapid Access to Polysubstituted Spirocyclic Hexahydroxanthones. Synthesis 2020, 52, 3047–3057. [Google Scholar]
- Zou, C.; Yang, L.; Zhang, L.; Liu, C.; Ma, Y.; Song, G.; Liu, Z.; Cheng, R.; Ye, J. Enantioselective Vinylogous Conia-Ene Reaction Catalyzed by a Disilver(I)/Bisdiamine Complex. ACS Catal. 2021, 11, 6865–6871. [Google Scholar] [CrossRef]
- Karimi, B.; Tavakolian, M.; Akbari, M.; Mansouri, F. Ionic Liquids in Asymmetric Synthesis: An Overall View from Reaction Media to Supported Ionic Liquid Catalysis. ChemCatChem 2018, 10, 3173–3205. [Google Scholar] [CrossRef]
- Singh, S.K.; Savoy, A.W. Ionic liquids synthesis and applications: An overview. J. Mol. Liq. 2020, 297, 112038. [Google Scholar] [CrossRef]
- Ahmadkhani, A.; Rad-Moghadam, K.; Roudsari, S.T. A Highly Enantioselective and Efficient Synthesis of New Pyrimidine-Fused Spiro[indoline-3,4’-pyran]s Promoted by a Novel Chiral Ionic Liquid. ChemistrySelect 2019, 4, 10442–10446. [Google Scholar] [CrossRef]
- Ji, Y.; Li, L.; Zhu, G.; Zhou, Y.; Lu, X.; He, W.; Gao, L.; Rong, L. Efficient reactions for the synthesis of pyrazolo[3,4-b] pyridine and pyrano[2,3-c]pyrazole derivatives from N-methyl-1-(methylthio)-2-nitroethen-1-amine. J. Heterocycl. Chem. 2020, 57, 1–16. [Google Scholar] [CrossRef]
- Tufail, F.; Saquib, M.; Singh, S.; Tiwari, J.; Dixit, P.; Singh, J.; Singh, J. A Practical Green Approach to Diversified Spirochromene/Spiropyran Scaffolds via a Glucose-Water Synergy Driven Organocatalytic System. New J. Chem. 2018, 42, 17279–17290. [Google Scholar] [CrossRef]
- Karimi-Jaberi, Z.; Fereydoonnezhad, A. One-pot, organocatalytic synthesis of spirooxindoles using citric acid in aqueous media. Iran. Chem. Commun. 2017, 5, 407–416. [Google Scholar]
- Mohamadpour, F.; Maghsoodlou, M.T.; Lashkari, M.; Heydari, R.; Hazeri, N. Synthesis of Quinolines, Spiro[4H-pyran-oxindoles] and Xanthenes Under Solvent-Free Conditions. Org. Prep. Proced. Int. 2019, 51, 456–476. [Google Scholar] [CrossRef]
- Li, M.-M.; Duan, C.-S.; Yu, Y.-Q.; Xu, D.-Z. A general and efficient one-pot synthesis of spiro[2-amino-4H-pyrans] via tandem multi-component reactions catalyzed by Dabco-based ionic liquids. Dyes Pigm. 2018, 150, 202–206. [Google Scholar] [CrossRef]
- Molla, A.; Ranjan, S.; Rao, M.S.; Dar, A.H.; Shyam, M.; Jayaprakash, V.; Hussain, S. Borax Catalysed Domino Synthesis of Highly Functionalised Spirooxindole and Chromenopyridine Derivatives: X-Ray Structure, Hirshfeld Surface Analysis and Molecular Docking Studies. ChemistrySelect 2018, 3, 8669–8677. [Google Scholar] [CrossRef]
- Saneinezhad, S.; Mohammadi, L.; Zadsirjan, V.; Bamoharram, F.F.; Heravi, M.M. Silver nanoparticles-decorated Preyssler functionalized cellulose biocomposite as a novel and efficient catalyst for the synthesis of 2-amino-4H-pyrans and spirochromenes. Sci. Rep. 2020, 10, 14540. [Google Scholar] [CrossRef] [PubMed]
- Kamali, F.; Shirini, F. An efficient one-pot multi-component synthesis of spirooxindoles using Fe3O4/g-C3N4 nanocomposite as a green and reusable catalyst in aqueous media. J. Mol. Struct. 2021, 1227, 129654. [Google Scholar] [CrossRef]
- Heravi, M.M.; Momeni, T.; Mirzaei, M.; Zadsirjan, V.; Tahmasebi, M. An amino acid@isopolyoxometalate nanoparticles catalyst containing aspartic acid and octamolybdate for the synthesis of functionalized spirochromenes. Inorg. Nano-Met. 2021, 51, 896–909. [Google Scholar] [CrossRef]
- Dadaei, M.; Naeimi, H. Guanidine functionalized core–shell structured magnetic cobalt-ferrite: An efficient nanocatalyst for sonochemical synthesis of spirooxindoles in water. RSC Adv. 2021, 11, 15360. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Gaber, A.E.-A.M.; Mcnab, H. Synthetic Applications of the Pyrolysis of Meldrum’s Acid Derivatives. Synthesis 2001, 14, 2059–2074. [Google Scholar] [CrossRef]
- Kamalifar, S.; Kiyani1, H. An Expeditious One-Pot Three-component Synthesis of 4-Aryl-3,4-dihydrobenzo[g]quinoline-2,5,10(1H)-triones under Green Conditions. Curr. Org. Chem. 2019, 23, 2626–2634. [Google Scholar] [CrossRef]
- Dourado, G.A.A. Reatividade da 2-Amino-1,4-naftoquinona em Reações de Hantzsch: Síntese Tricomponente de Azapolicíclos Funcionalizados. Master’s Thesis, Chemistry Department, Federal University of Bahia, Salvador, BA, Brazil, 2018; 120p. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Catalyst | Amount of Catalyst (mmol) | Yield (%) |
|---|---|---|---|---|
| 1 | CH3CN | IL | 0.3 | 81 |
| 2 | H2O | IL | 0.3 | 71 |
| 3 | Acetone | IL | 0.3 | 28 |
| 4 | CH2Cl2 | IL | 0.3 | 51 |
| 5 | Et2O | IL | 03 | 39 |
| 6 | Toluene | IL | 0.3 | 23 |
| 7 | EtOH | IL | 0.3 | 85 |
| 8 | EtOH | p-TsOH | 0.3 | 80 |
| 9 | EtOH | Glucose | 0.3 | 39 |
| 10 | EtOH | Citric acid | 0.3 | 67 |
| 11 | EtOH | TFA | 0.3 | 80 |
| 12 | EtOH | IL | 0.2 | 55 |
| 13 | EtOH | IL | 0.1 | 65 |
| 14 | EtOH | IL | 0.05 | 58 |
| Entry | Solvent | Heating | Temperature | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | EtOH | - | 0 °C | 34 | 36 |
| 2 | EtOH | - | r.t. | 72 | 71 |
| 3 | EtOH | Conventional | Reflux | 24 | 85 |
| 4 | EtOH | Microwave | 80 °C | 2 | 91 |
| Cell Lines—IC50 (μM) | ||||
|---|---|---|---|---|
| Compounds | HCT116 | PC3 | SNB19 | HL60 |
| 1a | >200 | >200 | >200 | >200 |
| 1b | 89.45 | 110.7 | 124.9 | 77.38 |
| 1c | 52.81 | 74.40 | 101.9 | 49.72 |
| 1d | >200 | >200 | >200 | >200 |
| 1e | 67.29 | 117.6 | >200 | 72.26 |
| 1f | 165.8 | >200 | >200 | 117.2 |
| 1g | >200 | >200 | >200 | 99.07 |
| 1h | >200 | >200 | >200 | >200 |
| 1i | >200 | >200 | >200 | >200 |
| 1j | >200 | >200 | >200 | >200 |
| 1k | >200 | >200 | >200 | >200 |
| 1l | >200 | >200 | >200 | >200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westphal, R.; Venturini Filho, E.; Loureiro, L.B.; Tormena, C.F.; Pessoa, C.; Guimarães, C.d.J.; Manso, M.P.; Fiorot, R.G.; Campos, V.R.; Resende, J.A.L.C.; et al. Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted. Molecules 2022, 27, 8051. https://doi.org/10.3390/molecules27228051
Westphal R, Venturini Filho E, Loureiro LB, Tormena CF, Pessoa C, Guimarães CdJ, Manso MP, Fiorot RG, Campos VR, Resende JALC, et al. Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted. Molecules. 2022; 27(22):8051. https://doi.org/10.3390/molecules27228051
Chicago/Turabian StyleWestphal, Regina, Eclair Venturini Filho, Laiza Bruzadelle Loureiro, Cláudio Francisco Tormena, Claudia Pessoa, Celina de Jesus Guimarães, Mariana Palmeira Manso, Rodolfo Goetze Fiorot, Vinicius Rangel Campos, Jackson Antônio Lamounier Camargos Resende, and et al. 2022. "Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted" Molecules 27, no. 22: 8051. https://doi.org/10.3390/molecules27228051
APA StyleWestphal, R., Venturini Filho, E., Loureiro, L. B., Tormena, C. F., Pessoa, C., Guimarães, C. d. J., Manso, M. P., Fiorot, R. G., Campos, V. R., Resende, J. A. L. C., Medici, F., & Greco, S. J. (2022). Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted. Molecules, 27(22), 8051. https://doi.org/10.3390/molecules27228051