1. Introduction
Protein therapeutics are new-generation drugs produced by the biopharmaceutical industry, utilizing the machinery of various cell lines during bioprocessing [
1,
2]. These recombinant therapeutic proteins, i.e., the products manufactured with these new technologies, are mostly monoclonal antibodies (mAb) [
3]. However, other therapeutic antibody-related products such as Fc-fusion proteins, antibody fragments and antibody–drug conjugates have also become a progressively important part of the biotherapeutics palette [
4] and utilized to treat a wide variety of medical conditions including paroxysmal nocturnal hemoglobinuria [
5], cryopyrin-associated periodic syndromes [
6], different types of cancer [
7], multiple sclerosis [
8], asthma and rheumatoid arthritis [
9], just to mention a few. Their widespread use is reflected in the current market dominance of biopharmaceutical drugs worldwide [
10]. Due to the significance of these biological drugs, understanding of their structure–function relationship is of high importance. In the case of glycoprotein therapeutics, one of the important critical quality attributes (CQA) is their N-glycosylation profile [
11]. Asparagine-linked carbohydrates have influences on antibody conformation, effector function, stability, solubility, secretion, pharmacokinetics and immunogenicity [
12]. The identification and characterization of the attached oligosaccharides are conducted in most instances at the unconjugated glycan level. PNGase F-mediated N-linked carbohydrate release is one of the most extensively used methods in such studies [
13,
14]. However, it has been reported that the released oligosaccharides and certain sugar ingredients in the sample may influence the endoglycosidase catalyzed reaction, e.g., glucose present in the formulation material [
15].
The kinetics of enzyme-mediated biochemical reactions are usually modeled by the Michaelis-Menten method, considering that the enzyme (E) forms an intermediate complex (ES) with the substrate (S), which is later converted to the product (P), while the enzyme is regenerated [
16]:
Therefore, the Michaelis-Menten concept can be readily utilized to study enzymatic reaction kinetics as follows:
where
V is the average reaction rate,
Vmax is the maximum reaction rate, [S] is the substrate concentration and K
M is the Michaelis-Menten constant. This latter (K
M) allows us to make relative comparisons between the kinetics of different enzymes with the same specificity [
17]. In the case of glycoprotein substrates, the reaction products can be rapidly quantified by sodium dodecyl sulfate capillary gel electrophoresis (SDS-CGE), a high-resolution and automated approach to SDS-PAGE that readily separates the deglycosylated and intact proteoforms [
18]. As a matter of fact, SDS-CGE is widely used in the biopharmaceutical industry to analyze therapeutic proteins in a simple and robust manner during drug development, production and release testing [
19]. Capillary isoelectric focusing (cIEF) is another frequently used bioanalytical method used to gain information about the charge heterogeneity of therapeutic proteins, therefore increasingly adopted in recent years for their characterization [
18].
2. Results
In this study, first, the digestion reaction kinetics of our in-house produced 6His-PNGase F [
20] was evaluated using human immunoglobulin-G1 (hIgG1) as substrate. The digestion products were analyzed by SDS-CGE to follow the reaction speed, and Michaelis-Menten plots were used to shed light on the release efficiency. The Michaelis-Menten constants and the maximum reaction rates were comparatively determined in the presence and absence of glucose in the digestion reaction mixture for a frequently used commercial endoglycosidase and the in-house produced 6His-PNGase F. Finally, capillary isoelectric focusing was employed to study the charge heterogeneity profiles of the two enzymes.
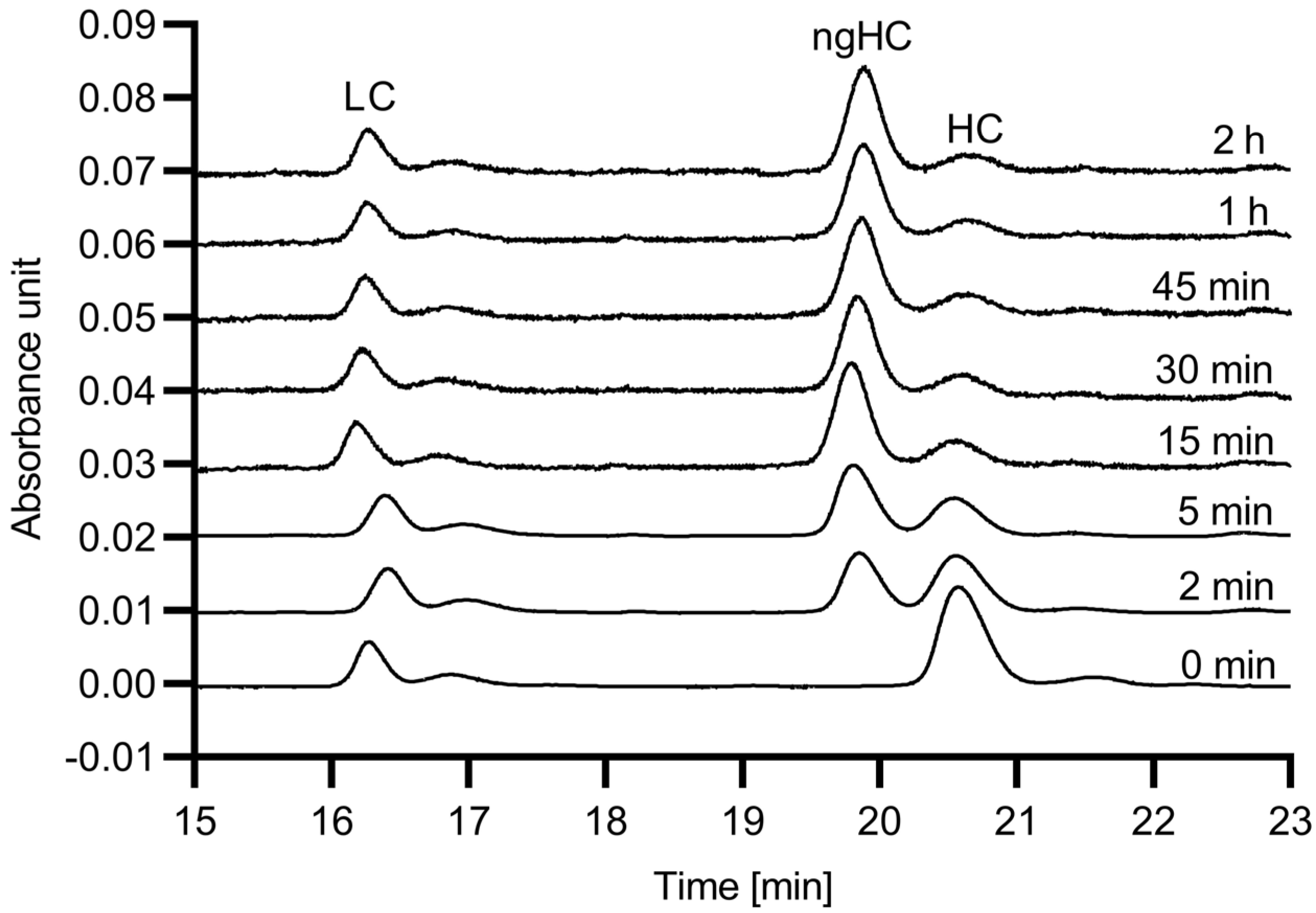
First, the digestion efficiency of the 6His-PNGase F enzyme was evaluated with respect to reaction speed using hIgG1 as substrate. The digestion reactions were conducted up to 120 min, and SDS-CGE was used to quantify the release efficiency, based on the resulting peak area changes for the glycosylated and non-glycosylated heavy chain fragments.
Figure 1 shows the obtained electropherograms.
As the endoglycosidase digestion reaction proceeded, the peak area of the glycosylated mAb heavy chain fragment (HC) decreased, while the peak representing the non-glycosylated heavy chain (ngHC) increased. Please note that almost full deglycosylation occurred within the first 15 min.
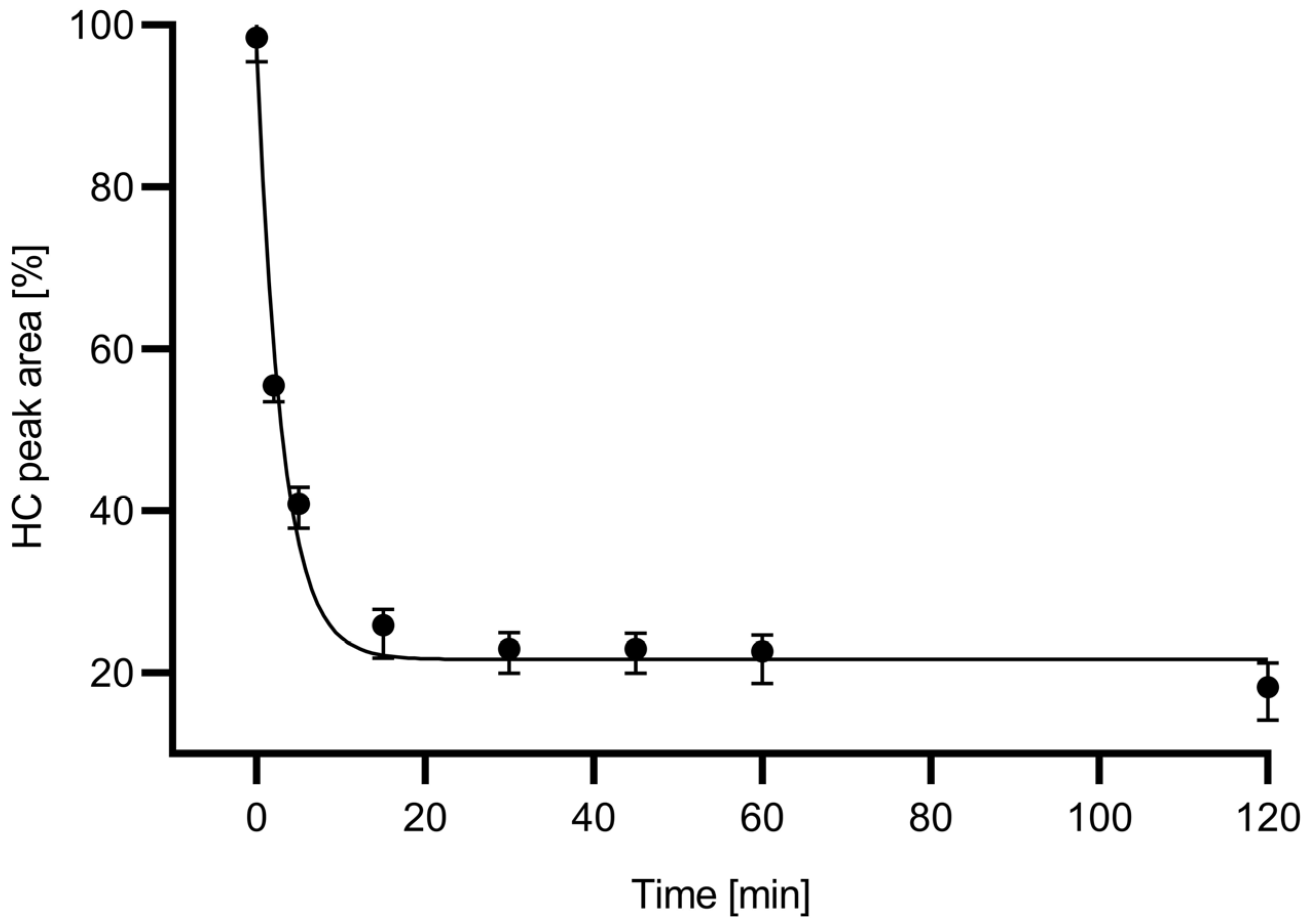
Figure 2 shows the digestion time plot based on the peak area decrease in the HC subunit with the use of the in-house produced 6His-PNGase F enzyme.
Based the deglycosylation time plot in
Figure 2, a 15 min digestion time was chosen for all downstream experiments because during this time frame, apparently close to maximum release rate was already achieved. The relative ngHC peak area % was calculated as depicted by Equation (3) and used accordingly in all further calculations.
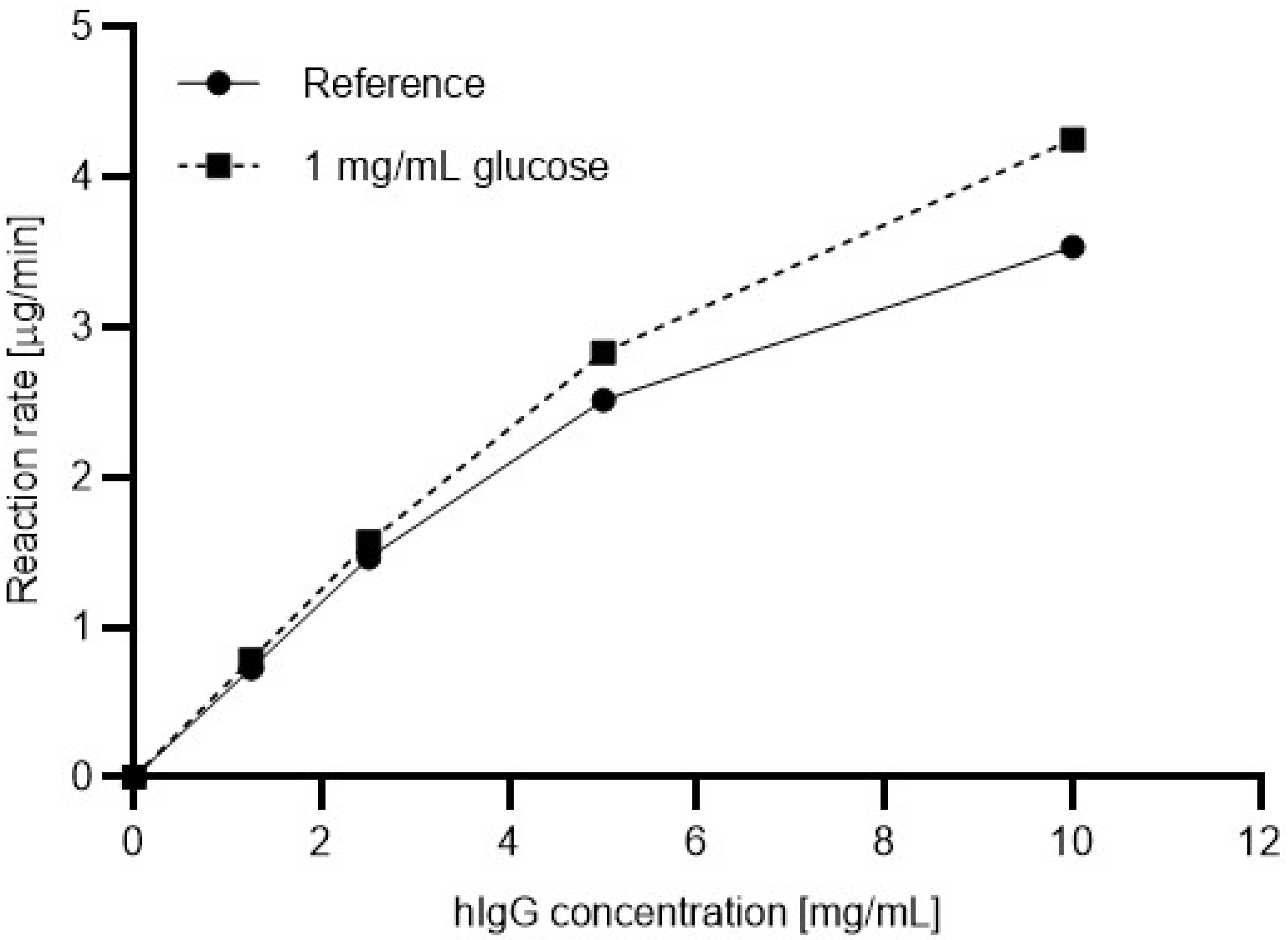
Next, the Michaelis-Menten plots were generated with the presence and absence of glucose in the reaction mixtures. Four different substrate concentrations were prepared ranging from 1.5 mg/mL to 10 mg/mL. Please note that the use of a substrate concentration higher than 10 mg/mL resulted in precipitation during the denaturation step. To understand the effect of the sugar content in the sample on the reaction kinetics, glucose was added to the reaction mixtures in 1 mg/mL final concentration.
Figure 3 shows the resulting Michaelis-Menten plots based on the relative ngHC peak area % values (Equation (3)), without (solid line) and with (dashed line) the presence of 1 mg/mL glucose in the digestion reaction using the in-house produced 6His-PNGase F. The reaction rates were determined as follows:
As one can observe, with increasing substrate concentration, the reaction rate increased in a higher degree with the presence of glucose in the reaction mixture. Apparently, with 10 mg/mL hIgG1 substrate concentration, the addition of 1 mg/mL glucose resulted in a 16.8% improvement in the reaction rate. Therefore, glucose should be considered as an activator for the 6His-PNGase F enzyme.
Based on the results shown in
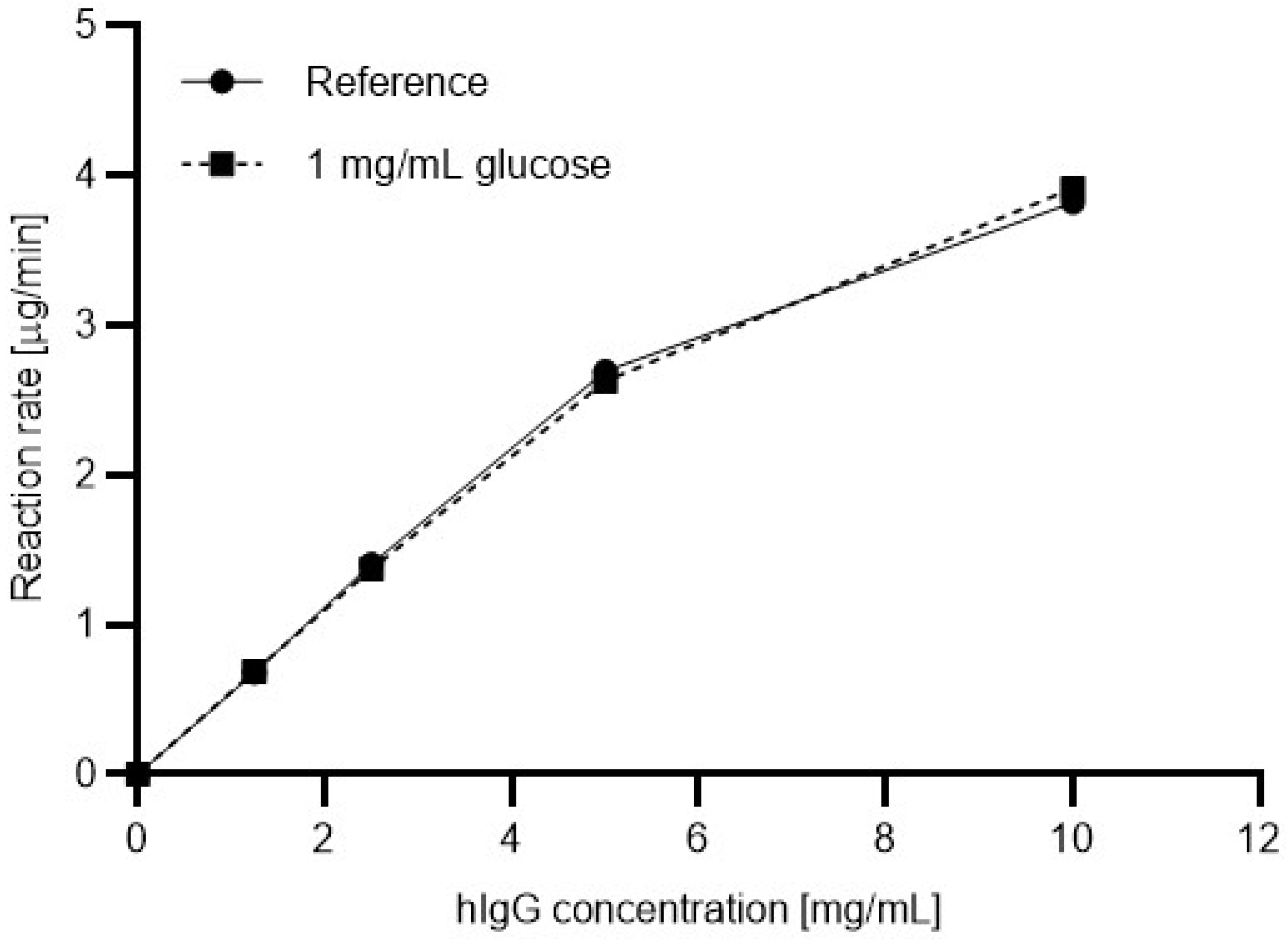
Figure 3, the digestion efficiency was also evaluated for a commonly used commercial PNGase F enzyme.
Figure 4 shows the resulting Michaelis-Menten plots for the commercial enzyme with (dashed line) and without (solid line, reference) 1 mg/mL glucose in the reaction mixture. In this instance, apparently no significant differences were found between the reaction rates.
For comparative purposes, the maximum reaction rates (
Vmax) and the Michaelis-Menten constants (K
M) were calculated from
Figure 3 and
Figure 4 for both enzymes with and without the presence of 1 mg/mL glucose in the reaction mixture using the GraphPad Prism software (GraphPad Software, San Diego, CA, USA). Please note that these values were predicted by the software since the precipitation of the IgG at a concentration higher than 10 mg/mL hampered further measurements. The significantly increasing reaction rate (activation) with the in-house produced 6His-PNGase F (UP) in the presence of 1 mg/mL glucose (
Figure 3) was in contrast to the less than moderate change with the use of the commercial enzyme (
Figure 4), as the corresponding maximum reaction rates and Michaelis-Menten constants depict in
Table 1.
As shown by the data in
Table 1, the
Vmax value of the commercial enzyme was 23% higher than that of the 6His-PNGase F without having glucose in the reaction mixture. On the other hand, when 1 mg/mL glucose was present in the digestion reaction, the 6His-PNGase F performed slightly (3%) better than its commercial counterpart with the application of the same enzyme units. More interestingly, the addition of glucose increased the reaction rate of the commercial enzyme only by 12% but showed significant activation (43%) in the performance of the 6His-PNGase F. The associated K
M values followed the same trend according to the Michaelis-Menten theory.
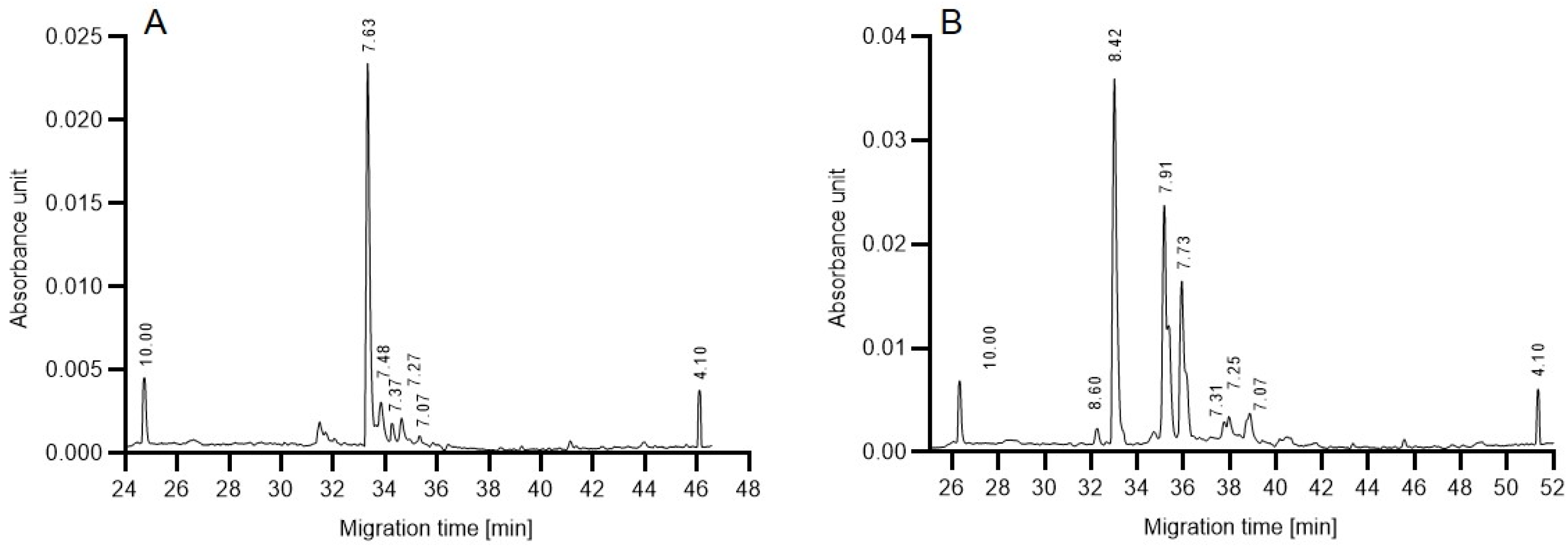
As a first step towards gaining some preliminary understanding of the above described reaction rate differences, isoelectric focusing (cIEF) was performed to compare the charge heterogeneities of the in-house produced 6His-PNGase F and the commercial enzyme. The resulting separation traces in
Figure 5 reveal subtle differences between the profiles. Although the functional group of both endoglycosidases is assumed to be similar, the in-house produced 6His-PNGase F enzyme (
Figure 5A) featured only one dominant peak at pH 7.63 with no significant acidic and basic variants. Please note that the His-tag is known to shift the pI of the conjugate towards the acidic region. In the cIEF trace of the commercial enzyme (
Figure 5B), on the other hand, two major acidic isoforms appeared (pHs 7.91 and 7.73) in addition to the main peak at pH 8.42. As a first approximation, we consider that the presence and absence of these acidic variants may be somewhat responsible for the observed activity variations in the presence of glucose during the deglycosylation reaction.
4. Materials and Methods
4.1. Chemicals and Reagents
Sodium hydroxide, phosphoric acid, glacial acetic acid, urea, iminodiacetic acid, 2-mercaptoethanol and L-arginine were from Sigma Aldrich (St. Louis, MO, USA). The Pharmalyte 3–10 was from GE Healthcare (Chicago, IL, USA). The cIEF Gel; the pI markers of the pI Peptide Marker kit; the Fast Glycan kit; and the SDS-MW Analysis Assay kit including the SDS-MW Gel Buffer, Sample Buffer and 10 kDa protein standard along with the 50 µm ID NCHO and BFS capillaries were from Bio-Science Kft (Budapest, Hungary). The hIgG1 test sample was from Molecular Innovations (Novi, MI, USA). The commercial PNGase F enzyme was from New England Biolabs (Ipswich, MA, USA). The in-house produced 6His-PNGase F (production described in detail in [
20] and this enzyme featured 3 months of shelf life) was from University of Pannonia (Veszprem, Hungary).
4.2. PNGase F Digestion
During the enzyme kinetic study, 1.5–10 µL of 10 mg/mL hIgG1 was used as the substrate. First, the sample was diluted to 10 µL (if necessary) and denatured by the addition of 2.0 µL of a denaturation mixture (Fast Glycan Kit, Bio-Science Kft, Budapest, Hungary) followed by incubation at 70 °C for 15 min. Then, 20 µL of 18 mM ammonium acetate was added to the denatured samples, and the N-glycans were released by the addition of 1 µL of the 6His-tagged or the commercial PNGase F enzyme (1.5 mU). The reaction mixture was incubated at 37 °C for the time periods shown in the corresponding parts of the manuscript. The endoglycosidase digestion reaction was stopped by the addition of the SDS-MW denaturation buffer.
4.3. Sample Preparation for SDS-CGE and cIEF
For reduced protein sample preparation, 80 µL of SDS-MW Sample Buffer was mixed with 5.0 µL of 2-mercaptoethanol, 2.0 µL of 10 kDa marker and 10 µL of the undigested or PNGase F digested (each individual time points) glycoprotein solutions followed by incubation at 70 °C for 15 min in a PCR instrument with heated lid.
The cIEF Master Mix contained the cIEF gel, 3.75 M urea, 48 mL of Pharmalyte 3–10, 80 mL of 500 mM L-arginine, 8.0 mL of 200 mM iminodiacetic acid, 1-1 mL of pI Marker 4.1 and 10.0 each; 100 µL of Master Mix and 2.0 µL of 10 mg/mL sample protein were mixed prior to the cIEF measurements.
4.4. SDS-CGE Analysis
An MDQ Capillary Electrophoresis System (Beckman Coulter, Fullerton, CA, USA) was used with UV detection (220 nm) for all SDS-CGE analyses, employing the SDS-MW Gel Buffer in a 20 cm effective length (30 cm total length), 50 μm ID bare fused capillary. The column was conditioned prior to each measurement and between runs by rinsing for 5 min each with HPLC grade water, 0.5 M NaOH and 0.5 M HCl. The separation voltage was set to 15 kV in reverse polarity mode (cathode at the injection side). The samples were injected by applying 5.0 kV for 20 s, also in reverse polarity mode. The separation temperature was 25 °C. For data acquisition and analysis, the 32Karat (version 8.0) software package (Beckman Coulter, Fullerton, CA, USA) was used. The GraphPad Prism (8.0.1 version, GraphPad Software, San Diego, CA, USA) software was used for the calculation of Vmax and KM values.
4.5. cIEF Analysis
The same capillary electrophoresis system was used for all cIEF measurements but with UV detection at 280 nm. For the separations, a 50 µM ID, 30 cm total length (20 cm effective length) NHCO capillary was employed. The sample storage compartment was set to 10 °C. Prior to injections; the capillary was washed with 6 M urea at 50 psi for 5.0 min followed by HPLC grade water at 50 psi for 5.0 min. The injection was achieved by filling up the capillary with the sample, mixed into the Master Mixture (25 psi/100 s). The formation of the pH gradient and focusing started by applying an 833 V/cm electric field in normal polarity mode for 15 min between the anolyte (200 mM phosphoric acid) and catholyte (300 mM sodium hydroxide) solutions. Then, the mobilization was performed using 1000 V/cm electric field (30 kV) in normal polarity mode between the anolyte and the chemical mobilizer (350 mM acetic acid) for 45 min at 20 °C capillary cartridge temperature. Data acquisition and analysis were completed as described above.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}