
Exploratory Metabolomics Underscores the Folate Enzyme ALDH1L1 as a Regulator of Glycine and Methylation Reactions

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Knockout of ALDH1L1 in RT4 Cells
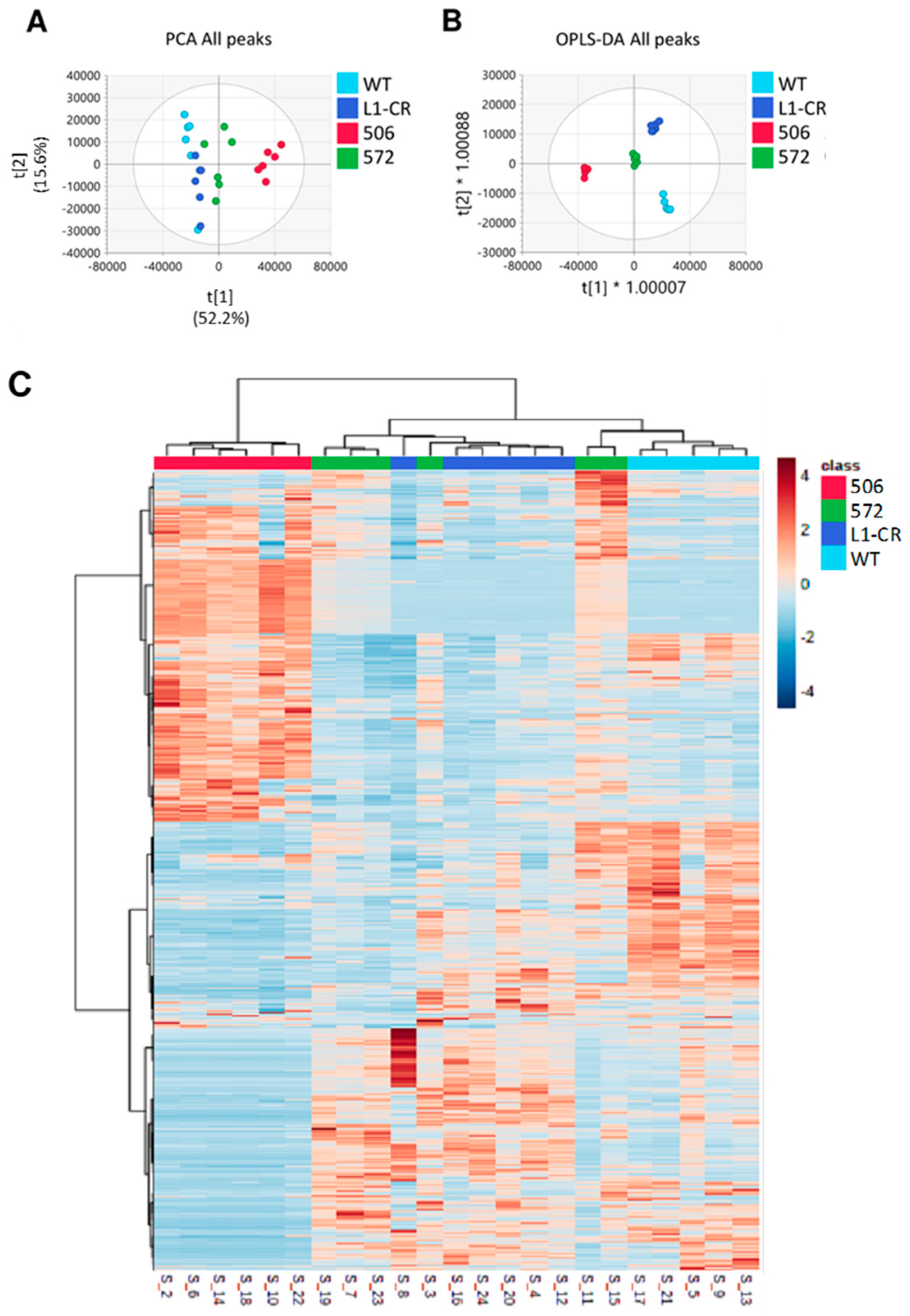
2.2. Overall Metabolomic Analysis
2.3. Top Metabolites Separating RT4 Cells and ALDH1L1-Deficient Clones
2.4. Metabolic Differences between RT4 Cells with High, Low, and Undetectable Levels of ALDH1l1
2.5. Construction of ALDH1L1-Dependent Metabolic Network
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Aldh1l1-Knockout Cell Lines
4.3. Western Blot Assays
4.4. Immunofluorescence Staining
4.5. Metabolite Extraction
4.6. Ultra High Performance Liquid Chromatography-High Resolution Mass Spectrometry (UHPLC-HRMS) Analysis
4.7. Metabolite Identification/Annotation and Metabolite Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Krupenko, S.A.; Oleinik, N.V. 10-Formyltetrahydrofolate Dehydrogenase, One of The Major Folate Enzymes, Is Down-Regulated In Tumor Tissues and Possesses Suppressor Effects On Cancer Cells. Cell Growth Differ. 2002, 13, 227–236. [Google Scholar]
- Krupenko, S.A. Fdh: An Aldehyde Dehydrogenase Fusion Enzyme In Folate Metabolism. Chem. Biol. Interact. 2009, 178, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Krupenko, S.A.; Krupenko, N.I. Loss of Aldh1l1 Folate Enzyme Confers A Selective Metabolic Advantage For Tumor Progression. Chem. Biol. Interact. 2019, 302, 149–155. [Google Scholar] [CrossRef]
- Anguera, M.C.; Field, M.S.; Perry, C.; Ghandour, H.; Chiang, E.P.; Selhub, J.; Shane, B.; Stover, P.J. Regulation of Folate-Mediated One-Carbon Metabolism By 10-Formyltetrahydrofolate Dehydrogenase. J. Biol. Chem. 2006, 281, 18335–18342. [Google Scholar] [CrossRef] [Green Version]
- Hoeferlin, L.A.; Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Activation of P21-Dependent G1/G2 Arrest In The Absence of Dna Damage As An Antiapoptotic Response To Metabolic Stress. Genes Cancer 2011, 2, 889–899. [Google Scholar] [CrossRef] [Green Version]
- Krupenko, N.I.; Sharma, J.; Pediaditakis, P.; Fekry, B.; Helke, K.L.; Du, X.; Sumner, S.; Krupenko, S.A. Cytosolic 10-Formyltetrahydrofolate Dehydrogenase Regulates Glycine Metabolism In Mouse Liver. Sci. Rep. 2019, 9, 14937. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Rushing, B.R.; Hall, M.S.; Helke, K.L.; Mcritchie, S.L.; Krupenko, N.I.; Sumner, S.J.; Krupenko, S.A. Sex-Specific Metabolic Effects of Dietary Folate Withdrawal In Wild-Type and Aldh1l1 Knockout Mice. Metabolites 2022, 12, 454. [Google Scholar] [CrossRef]
- Krupenko, S.A.; Cole, S.A.; Hou, R.; Haack, K.; Laston, S.; Mehta, N.R.; Comuzzie, A.G.; Butte, N.F.; Voruganti, V.S. Genetic Variants In Aldh1l1 and Gldc Influence Serine To Glycine Ratio In Hispanic Children. Am. J. Clin. Nutr. 2022, 116, 500–510. [Google Scholar] [CrossRef]
- Shane, B.; Stokstad, E.L. Vitamin B12-Folate Interrelationships. Annu. Rev. Nutr. 1985, 5, 115–141. [Google Scholar] [CrossRef]
- Cooperman, J.M.; Lopez, R. The Role of Histidine In the Anemia of Folate Deficiency. Exp. Biol. Med. 2002, 227, 998–1000. [Google Scholar] [CrossRef]
- Beniaminov, A.D.; Puzanov, G.A.; Krasnov, G.S.; Kaluzhny, D.N.; Kazubskaya, T.P.; Braga, E.A.; Kudryavtseva, A.V.; Melnikova, N.V.; Dmitriev, A.A. Deep Sequencing Revealed A Cpg Methylation Pattern Associated With Aldh1l1 Suppression In Breast Cancer. Front. Genet. 2018, 9, 169. [Google Scholar] [CrossRef]
- Tackels-Horne, D.; Goodman, M.D.; Williams, A.J.; Wilson, D.J.; Eskandari, T.; Vogt, L.M.; Boland, J.F.; Scherf, U.; Vockley, J.G. Identification of Differentially Expressed Genes In Hepatocellular Carcinoma and Metastatic Liver Tumors By Oligonucleotide Expression Profiling. Cancer 2001, 92, 395–405. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, R.; Li, L.; Mao, L.; Liu, S.; Qiao, S.; Ren, G.; Hu, J. Validation of Gene Profiles For Analysis of Regional Lymphatic Metastases In Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2020, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, A.A.; Kashuba, V.I.; Haraldson, K.; Senchenko, V.N.; Pavlova, T.V.; Kudryavtseva, A.V.; Anedchenko, E.A.; Krasnov, G.S.; Pronina, I.V.; Loginov, V.I.; et al. Genetic and Epigenetic Analysis of Non-Small Cell Lung Cancer With Noti-Microarrays. Epigenetics 2012, 7, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.J.; Giannini, C.; Asmann, Y.W.; Sharma, M.K.; Perry, A.; Tibbetts, K.M.; Jenkins, R.B.; Scheithauer, B.W.; Anant, S.; Jenkins, S.; et al. Gene Expression Profiling of Nf-1-Associated and Sporadic Pilocytic Astrocytoma Identifies Aldehyde Dehydrogenase 1 Family Member L1 (Aldh1l1) As An Underexpressed Candidate Biomarker In Aggressive Subtypes. J. Neuropathol. Exp. Neurol. 2008, 67, 1194–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Epigenetic Silencing of Aldh1l1, A Metabolic Regulator of Cellular Proliferation, In Cancers. Genes Cancer 2011, 2, 130–139. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Rudenko, E.E.; Kudryavtseva, A.V.; Krasnov, G.S.; Gordiyuk, V.V.; Melnikova, N.V.; Stakhovsky, E.O.; Kononenko, O.A.; Pavlova, L.S.; Kondratieva, T.T.; et al. Epigenetic Alterations of Chromosome 3 Revealed By Noti-Microarrays In Clear Cell Renal Cell Carcinoma. Biomed Res. Int. 2014, 2014, 735292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senchenko, V.N.; Kisseljova, N.P.; Ivanova, T.A.; Dmitriev, A.A.; Krasnov, G.S.; Kudryavtseva, A.V.; Panasenko, G.V.; Tsitrin, E.B.; Lerman, M.I.; Kisseljov, F.L.; et al. Novel Tumor Suppressor Candidates On Chromosome 3 Revealed By Noti-Microarrays In Cervical Cancer. Epigenetics 2013, 8, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Prakasam, A.; Ghose, S.; Oleinik, N.V.; Bethard, J.R.; Peterson, Y.K.; Krupenko, N.I.; Krupenko, S.A. Jnk1/2 Regulate Bid By Direct Phosphorylation At Thr59 In Response To Aldh1l1. Cell Death Dis. 2014, 5, E1358. [Google Scholar] [CrossRef]
- Oleinik, N.V.; Krupenko, S.A. Ectopic Expression of 10-Formyltetrahydrofolate Dehydrogenase In A549 Cells Induces G(1) Cell Cycle Arrest and Apoptosis. Mol. Cancer Res. 2003, 1, 577–588. [Google Scholar] [PubMed]
- Oleinik, N.V.; Krupenko, N.I.; Priest, D.G.; Krupenko, S.A. Cancer Cells Activate P53 In Response To 10-Formyltetrahydrofolate Dehydrogenase Expression. Biochem. J. 2005, 391, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.; Jeronimo, C.; Henrique, R.; Belo, L.; De Lourdes Bastos, M.; De Pinho, P.G.; Carvalho, M. Biomarkers In Bladder Cancer: A Metabolomic Approach Using In Vitro and Ex Vivo Model Systems. Int. J. Cancer 2016, 139, 256–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Tan, Q.; Geddie, W.R.; Jewett, M.A.; Phillips, N.; Ke, D.; Simmons, C.A.; Sun, Y. Biophysical Characterization of Bladder Cancer Cells With Different Metastatic Potential. Cell Biochem. Biophys. 2014, 68, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Iliou, A.; Panagiotakis, A.; Giannopoulou, A.F.; Benaki, D.; Kosmopoulou, M.; Velentzas, A.D.; Tsitsilonis, O.E.; Papassideri, I.S.; Voutsinas, G.E.; Konstantakou, E.G.; et al. Malignancy Grade-Dependent Mapping of Metabolic Landscapes In Human Urothelial Bladder Cancer: Identification of Novel, Diagnostic, and Druggable Biomarkers. Int. J. Mol. Sci. 2020, 21, 1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, S.A.; Sharma, J. Is ALDH1L1 Elevated in Lung Cancer? Comment on: Lee, S.-H.; et al. “The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer” Cancers 2020, 12, 1382. Cancers 2021, 13, 1691. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.E.; Li, S.; Xia, J. Metaboanalyst 5.0: Narrowing The Gap Between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. Nadph Homeostasis In Cancer: Functions, Mechanisms and Therapeutic Implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. Nadph Production By The Oxidative Pentose-Phosphate Pathway Supports Folate Metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative Flux Analysis Reveals Folate-Dependent Nadph Production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Deberardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, E1600200. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, K.; Vogl, F.C.; Ritter, A.P.; Zhu, W.; Nurnberger, N.; Kreutz, M.; Oefner, P.J.; Gronwald, W.; Gottfried, E. Distinct Metabolic Differences Between Various Human Cancer and Primary Cells. Electrophoresis 2013, 34, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Armitage, E.G.; Barbas, C. Metabolomics In Cancer Biomarker Discovery: Current Trends and Future Perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, T.; Song, X.; Huang, L.; Zang, Q.; Xu, J.; Bi, N.; Jiao, G.; Hao, Y.; Chen, Y.; et al. Spatially Resolved Metabolomics To Discover Tumor-Associated Metabolic Alterations. Proc. Natl. Acad. Sci. USA 2019, 116, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic Signatures of Cancer Cells and Stem Cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Fendt, S.M. The Metabolism of Cancer Cells During Metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, A.K.; Deberardinis, R.J. Applications of Metabolomics To Study Cancer Metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 2–14. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; Deberardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Tissue-Specific and Convergent Metabolic Transformation of Cancer Correlates With Metastatic Potential and Patient Survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’sullivan, J.; Sheahan, K.; Cantley, L.C.; Vander Heiden, M.G.; Vitkup, D. Heterogeneity of Tumor-Induced Gene Expression Changes In The Human Metabolic Network. Nat. Biotechnol. 2013, 31, 522–529. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. Nad Metabolic Dependency In Cancer Is Shaped By Gene Amplification and Enhancer Remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. Kras-Driven Metabolic Rewiring Reveals Novel Actionable Targets In Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [Green Version]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of Pkm2 In Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Krupenko, N.I.; Sharma, J.; Fogle, H.M.; Pediaditakis, P.; Strickland, K.C.; Du, X.; Helke, K.L.; Sumner, S.; Krupenko, S.A. Knockout of Putative Tumor Suppressor Aldh1l1 In Mice Reprograms Metabolism To Accelerate Growth of Tumors In A Diethylnitrosamine (Den) Model of Liver Carcinogenesis. Cancers 2021, 13, 3219. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeon, Y.; Kang, J.H.; Jang, H.; Lee, H.; Kim, S.Y. The Combination of Loss of Aldh1l1 Function and Phenformin Treatment Decreases Tumor Growth In Kras-Driven Lung Cancer. Cancers 2020, 12, 1382. [Google Scholar] [CrossRef]
- Verma, N.; Pink, M.; Boland, S.; Rettenmeier, A.W.; Schmitz-Spanke, S. Benzo[A]Pyrene-Induced Metabolic Shift From Glycolysis To Pentose Phosphate Pathway In The Human Bladder Cancer Cell Line Rt4. Sci. Rep. 2017, 7, 9773. [Google Scholar] [CrossRef]
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The Progression From A Lower To A Higher Invasive Stage of Bladder Cancer Is Associated With Severe Alterations In Glucose and Pyruvate Metabolism. Exp. Cell Res. 2015, 335, 91–98. [Google Scholar] [CrossRef]
- Loras, A.; Martinez-Bisbal, M.C.; Quintas, G.; Gil, S.; Martinez-Manez, R.; Ruiz-Cerda, J.L. Urinary Metabolic Signatures Detect Recurrences In Non-Muscle Invasive Bladder Cancer. Cancers 2019, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione Metabolism In Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.E.; Malats, N.; Rothman, N.; Real, F.X.; Kogevinas, M.; Karami, S.; Garcia-Closas, R.; Silverman, D.; Chanock, S.; Welch, R.; et al. Polymorphisms In One-Carbon Metabolism and Trans-Sulfuration Pathway Genes and Susceptibility To Bladder Cancer. Int. J. Cancer 2007, 120, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Stretch, C.; Eastman, T.; Mandal, R.; Eisner, R.; Wishart, D.S.; Mourtzakis, M.; Prado, C.M.; Damaraju, S.; Ball, R.O.; Greiner, R.; et al. Prediction of Skeletal Muscle and Fat Mass In Patients With Advanced Cancer Using A Metabolomic Approach. J. Nutr. 2012, 142, 14–21. [Google Scholar] [CrossRef]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The Chemistry of The Vitamin B3 Metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Landgraf, B.J.; Mccarthy, E.L.; Booker, S.J. Radical S-Adenosylmethionine Enzymes In Human Health and Disease. Annu. Rev. BioChem. 2016, 85, 485–514. [Google Scholar] [CrossRef]
- Mpanga, A.Y.; Siluk, D.; Jacyna, J.; Szerkus, O.; Wawrzyniak, R.; Markuszewski, M.; Matuszewski, M.; Kaliszan, R.; Markuszewski, M.J. Targeted Metabolomics In Bladder Cancer: From Analytical Methods Development and Validation Towards Application To Clinical Samples. Anal. Chim. Acta 2018, 1037, 188–199. [Google Scholar] [CrossRef]
- Saad, A.A.; O’connor, P.J.; Mostafa, M.H.; Metwalli, N.E.; Cooper, D.P.; Margison, G.P.; Povey, A.C. Bladder Tumor Contains Higher N7-Methylguanine Levels In Dna Than Adjacent Normal Bladder Epithelium. Cancer Epidemiol. Biomark. PRev. 2006, 15, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Rinne, M.L.; He, Y.; Pachkowski, B.F.; Nakamura, J.; Kelley, M.R. N-Methylpurine Dna Glycosylase Overexpression Increases Alkylation Sensitivity By Rapidly Removing Non-Toxic 7-Methylguanine Adducts. Nucleic Acids Res. 2005, 33, 2859–2867. [Google Scholar] [CrossRef]
- Nilov, D.; Maluchenko, N.; Kurgina, T.; Pushkarev, S.; Lys, A.; Kutuzov, M.; Gerasimova, N.; Feofanov, A.; Svedas, V.; Lavrik, O.; et al. Molecular Mechanisms of Parp-1 Inhibitor 7-Methylguanine. Int. J. Mol. Sci. 2020, 21, 2159. [Google Scholar] [CrossRef] [Green Version]
- Billson, H.A.; Harrison, K.L.; Lees, N.P.; Hall, C.N.; Margison, G.P.; Povey, A.C. Dietary Variables Associated With Dna N7-Methylguanine Levels and O6-Alkylguanine Dna-Alkyltransferase Activity In Human Colorectal Mucosa. Carcinogenesis 2009, 30, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Petrella, G.; Ciufolini, G.; Vago, R.; Cicero, D.O. The Interplay Between Oxidative Phosphorylation and Glycolysis As A Potential Marker of Bladder Cancer Progression. Int. J. Mol. Sci. 2020, 21, 8107. [Google Scholar] [CrossRef] [PubMed]
- Khan, Q.A.; Pediaditakis, P.; Malakhau, Y.; Esmaeilniakooshkghazi, A.; Ashkavand, Z.; Sereda, V.; Krupenko, N.I.; Krupenko, S.A. Chip E3 Ligase Mediates Proteasomal Degradation of The Proliferation Regulatory Protein Aldh1l1 During The Transition of Nih3t3 Fibroblasts From G0/G1 To S-Phase. PLoS ONE 2018, 13, E0199699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleinik, N.V.; Krupenko, N.I.; Reuland, S.N.; Krupenko, S.A. Leucovorin-Induced Resistance Against Fdh Growth Suppressor Effects Occurs Through Dhfr Up-Regulation. Biochem. Pharmacol. 2006, 72, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Schroder, M.; Sumner, S.C.J. Comparison of Lysis and Detachment Sample Preparation Methods For Cultured Triple-Negative Breast Cancer Cells Using Uhplc-Hrms-Based Metabolomics. Metabolites 2022, 12, 168. [Google Scholar] [CrossRef]
- Rushing, B.R.; Tilley, S.; Molina, S.; Schroder, M.; Sumner, S. Commonalities In Metabolic Reprogramming Between Tobacco Use and Oral Cancer. Int. J. Environ. Res. Public Health 2022, 19, 10261. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Rushing, B.R.; Harris, S.E.; Mcritchie, S.L.; Dominguez, D.; Sumner, S.J.; Dohlman, H.G. Multi-Omics Analysis of Multiple Glucose-Sensing Receptor Systems In Yeast. Biomolecules 2022, 12, 175. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Rushing, B.R.; Harris, S.E.; Mcritchie, S.L.; Jones, J.C.; Dominguez, D.; Sumner, S.J.; Dohlman, H.G. Multi-Omics Analysis of Glucose-Mediated Signaling By A Moonlighting Gbeta Protein Asc1/Rack1. PLoS Genet. 2021, 17, E1009640. [Google Scholar] [CrossRef]
- Valikangas, T.; Suomi, T.; Elo, L.L. A Systematic Evaluation of Normalization Methods In Quantitative Label-Free Proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and Considerations For The Use of System Suitability and Quality Control Samples In Mass Spectrometry Assays Applied In Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Tarcea, V.G.; Karnovsky, A.; Mirel, B.R.; Weymouth, T.E.; Beecher, C.W.; Cavalcoli, J.D.; Athey, B.D.; Omenn, G.S.; Burant, C.F.; et al. Metscape: A Cytoscape Plug-In For Visualizing and Interpreting Metabolomic Data In The Context of Human Metabolic Networks. Bioinformatics 2010, 26, 971–973. [Google Scholar] [CrossRef]
- Strickland, K.C.; Krupenko, N.I.; Krupenko, S.A. Molecular Mechanisms Underlying The Potentially Adverse Effects of Folate. Clin. Chem. Lab. Med. 2013, 51, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Vrieling, A.; Bueno-De-Mesquita, H.B.; Ros, M.M.; Kampman, E.; Aben, K.K.; Buchner, F.L.; Jansen, E.H.; Roswall, N.; Tjonneland, A.; Boutron-Ruault, M.C.; et al. One-Carbon Metabolism Biomarkers and Risk of Urothelial Cell Carcinoma In The European Prospective Investigation Into Cancer and Nutrition. Int. J. Cancer 2019, 145, 2349–2359. [Google Scholar] [CrossRef]
- Eich, M.L.; Rodriguez Pena, M.D.C.; Chandrashekar, D.S.; Chaux, A.; Agarwal, S.; Gordetsky, J.B.; Ferguson, J.E.; Sonpavde, G.P.; Netto, G.J.; Varambally, S. Expression and Role of Methylenetetrahydrofolate Dehydrogenase 1 Like (Mthfd1l) In Bladder Cancer. Transl. Oncol. 2019, 12, 1416–1424. [Google Scholar] [CrossRef]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmstrom, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder Cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rushing, B.R.; Fogle, H.M.; Sharma, J.; You, M.; McCormac, J.P.; Molina, S.; Sumner, S.; Krupenko, N.I.; Krupenko, S.A. Exploratory Metabolomics Underscores the Folate Enzyme ALDH1L1 as a Regulator of Glycine and Methylation Reactions. Molecules 2022, 27, 8394. https://doi.org/10.3390/molecules27238394
Rushing BR, Fogle HM, Sharma J, You M, McCormac JP, Molina S, Sumner S, Krupenko NI, Krupenko SA. Exploratory Metabolomics Underscores the Folate Enzyme ALDH1L1 as a Regulator of Glycine and Methylation Reactions. Molecules. 2022; 27(23):8394. https://doi.org/10.3390/molecules27238394
Chicago/Turabian StyleRushing, Blake R., Halle M. Fogle, Jaspreet Sharma, Mikyoung You, Jonathan P. McCormac, Sabrina Molina, Susan Sumner, Natalia I. Krupenko, and Sergey A. Krupenko. 2022. "Exploratory Metabolomics Underscores the Folate Enzyme ALDH1L1 as a Regulator of Glycine and Methylation Reactions" Molecules 27, no. 23: 8394. https://doi.org/10.3390/molecules27238394