


Understanding the Regioselectivity and the Molecular Mechanism of [3 + 2] Cycloaddition Reactions between Nitrous Oxide and Conjugated Nitroalkenes: A DFT Computational Study


Abstract
:1. Introduction
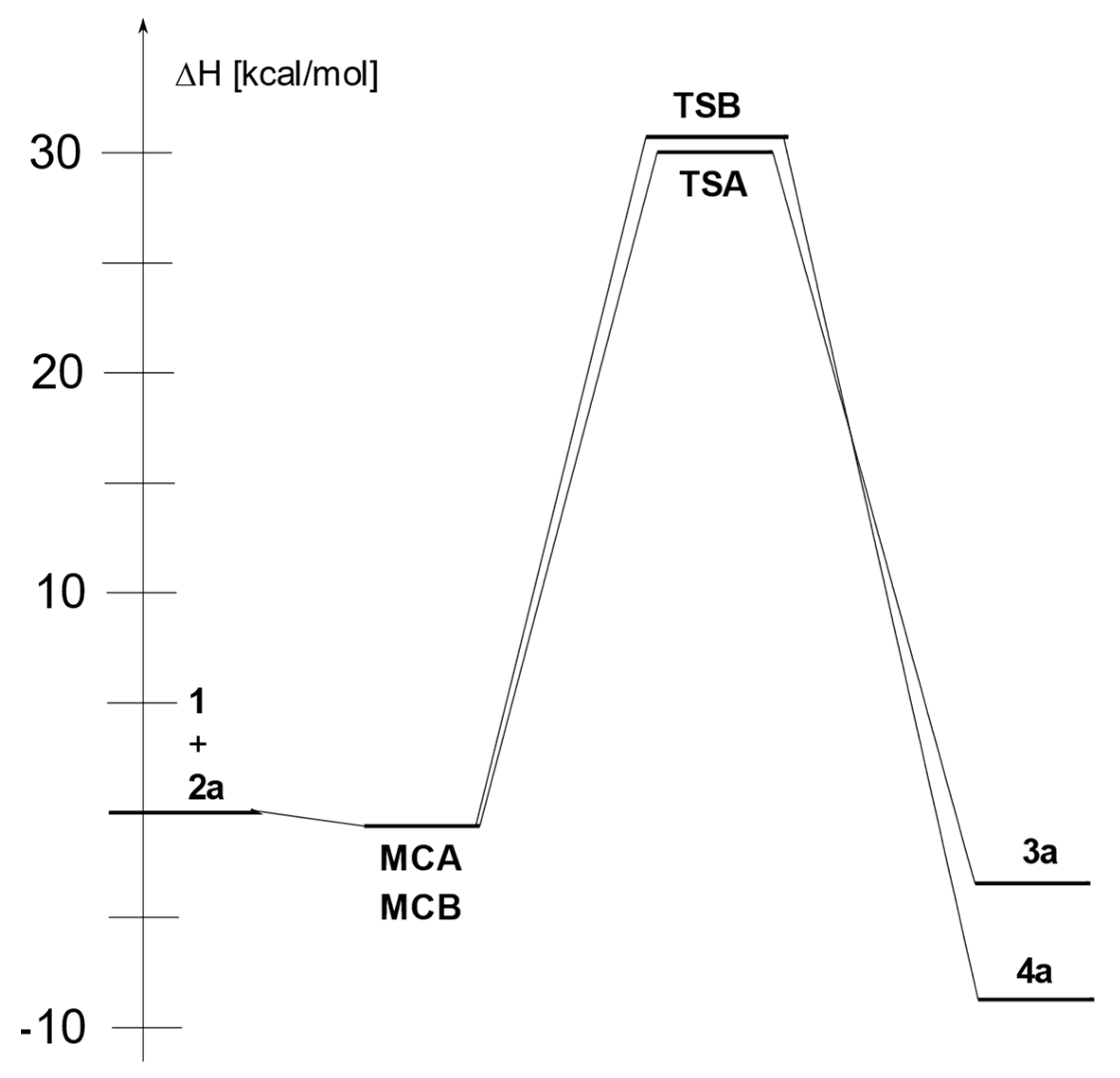
2. Results and Discussion
3. Computational Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Luo, L.; Wang, Q.; Xiang, Y.; Peng, X.; Hu, C. Synthesis and biological evaluation of novel thiazolo[4,5-d]pyrimidin-7(6H)-ones as topoisomerase I inhibitors. Chem. Heterocycl. Compd. 2021, 57, 1220–1229. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Ngo, T.H.; Tran, H.T.; Dinh, T.P.; Do, P.T.; Nguyen, H.B.; Tran, L.T.P.; Ta, H.M. Synthesis and Anticancer Activity of 11-azaartemisinin Derivatives Bearing 1,2,3-triazole Moiety. Chem. Heterocycl. Compd. 2021, 57, 1037–1044. [Google Scholar] [CrossRef]
- Bhat, S.I.; Kigga, M.; Heravi, M.M. Multicomponent reactions based on in situ generated isocyanides for the construction of heterocycles. Chem. Heterocycl. Compd. 2021, 57, 709–719. [Google Scholar] [CrossRef]
- Noriega, S.; Cardoso-Ortiz, J.; López-Luna, A.; Cuevas-Flores, M.D.R.; Flores De La Torre, J.A. The Diverse Biological Activity of Recently Synthesized Nitro Compounds. Pharmaceuticals 2022, 15, 717. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Comppounds to Amines. Org. Process Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Jasiński, R. Understanding the molecular mechanism of γ-elimination of nitrous acid in the framework of the molecular electron density theory. J. Comput. Chem. 2021, 42, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhang, S.; Zhao, Y.; Wang, D.Z. New Approach to Oximes through Reduction of Nitro Compounds Enabled by Visible Light Photoredox Catalysis. Org. Lett. 2013, 15, 2660–2663. [Google Scholar] [CrossRef]
- Kornblum, N.; Brown, R.A. The Synthesis and Characterization of Nitronic Esters. J. Am. Chem. Soc. 1964, 86, 2681–2687. [Google Scholar] [CrossRef]
- Hao, F.; Nishiwaki, N. Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules 2020, 25, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiński, R. A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett. 2015, 56, 532–535. [Google Scholar] [CrossRef]
- Fryźlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O.M.; Dresler, E.; Jasiński, R. Regio- and stereoselective synthesis of nitrofunctionalized 1,2-oxazolidine analogs of nicotine. Chem. Heterocycl. Compd. 2020, 56, 120–122. [Google Scholar] [CrossRef]
- Żmigrodzka, M.; Dresler, E.; Hordyjewicz-Baran, Z.; Kulesza, R.; Jasiński, R. A unique example of noncatalyzed [3+2] cycloaddition involving (2E)-3-aryl-2-nitroprop-2-enenitriles. Chem. Heterocycl. Compd. 2017, 53, 1161–1162. [Google Scholar] [CrossRef]
- Jasiński, R. In the searching for zwitterionic intermediates on reaction paths of [3 + 2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins. RSC Adv. 2015, 5, 101045–101048. [Google Scholar] [CrossRef]
- Fryźlewicz, A.; Kącka-Zych, A.; Demchuk, O.M.; Mirosław, B.; Woliński, P.; Jasiński, R. Green synthesis of nitrocyclopropane-type precursors of inhibitors for the maturation of fruits and vegetables via domino reactions of diazoalkanes with 2-nitroprop-1-ene. J. Clean. Prod. 2021, 292, 126079. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. The Participation of 3,3,3-Trichloro-1-nitroprop-1-ene in the [3+2] Cycloaddition Reaction with Selected Nitrile N-Oxides in the Light of the Experimental and MEDT Quantum Chemical Study. Molecules 2021, 26, 6774. [Google Scholar] [CrossRef] [PubMed]
- Woliński, P.; Kącka-Zych, A.; Demchuk, O.M.; Łapczuk-Krygier, A.; Mirosław, B.; Jasiński, R. Clean and molecularly programmable protocol for preparation of bis-heterobiarylic systems via a domino pseudocyclic reaction as a valuable alternative for TM-catalyzed cross-couplings. J. Clean. Prod. 2020, 275, 122086. [Google Scholar] [CrossRef]
- Jasiński, R. Nitroacetylene as dipolarophile in [2+3] cycloaddition reactions with allenyl-type three-atom components: DFT computational study. Monatsh. Chem. 2015, 146, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 2019, 267–282. [Google Scholar] [CrossRef]
- Jasiński, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review. Organics 2020, 1, 5. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kula, K.; Zawadzińska, K. Local nucleophile-electrophile interactions in [3+2] cycloaddition reactions between benzonitrile N-oxide and selected conjugated nitroalkenes in the light of MEDT computational study. Curr. Chem. Lett. 2021, 10, 9–16. [Google Scholar] [CrossRef]
- Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Jasiński, R. Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes. Pure Appl. Chem. 2021, 93, 427–446. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Pérez, P. Perfluorobicyclo[2.2.0]hex-1(4)-ene as unique partner for Diels–Alder reactions with benzene: A density functional theory study. Theor. Chem. Acc. 2021, 140, 17. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Understanding the uniqueness of the stepwise [4+1]cycloaddition reaction between conjugated nitroalkenesand electrophilic carbene systems with a molecular electrondensity theory perspective. Int. J. Quantum Chem. 2021, 121, 26440. [Google Scholar] [CrossRef]
- Kacka-Zych, A. Push-pull nitronates in the [3+2] cycloaddition with nitroethylene: Molecular Electron Density Theory study. J Mol. Graph. Model. 2020, 97, 1075492. [Google Scholar] [CrossRef]
- Kącka-Zych, A. The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3+2) Cycloaddition: A DFT Mechanistic Study. Molecules 2021, 26, 4786. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R. Stepwise, zwitterionic course of hetero-Diels–Alder reaction between 1,2,4-triazine molecular systems and 2-cyclopropylidene-1,3-dimethylimidazoline. Chem. Heterocycl. Comp. 2022, 58, 260–262. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ (eV) | η (eV) | ω (eV) | Δω (eV) | |
|---|---|---|---|---|
| 1 | −4.92 | 8.79 | 1.37 | |
| 2a | −5.33 | 5.45 | 2.61 | 1.23 |
| 2b | −5.16 | 5.48 | 2.43 | 1.05 |
| 2c | −5.98 | 5.03 | 3.56 | 2.18 |
| 2d | −5.08 | 5.48 | 2.35 | 0.98 |
| 2e | −6.49 | 4.66 | 4.52 | 3.15 |
| Solvent | Transition | ΔH | ΔS | ΔG |
|---|---|---|---|---|
| Toluene | 1 + 2a→ MCA | −0.2 | −14.1 | 4.0 |
| 1 + 2a→ TSA | 30.3 | −30.3 | 39.3 | |
| 1 + 2a→ 3a | −2.8 | −31.0 | 6.4 | |
| 1 + 2a→ MCB | −0.4 | −16.0 | 4.4 | |
| 1 + 2a→ TSB | 31.2 | −30.4 | 40.2 | |
| 1 + 2a→ 4a | −8.1 | −30.8 | 1.1 | |
| Acetone | 1 + 2a→ MCA | 0.0 | −11.5 | 3.5 |
| 1 + 2a→ TSA | 30.5 | −31.2 | 39.8 | |
| 1 + 2a→ 3a | −3.4 | −32.4 | 6.2 | |
| 1 + 2a→ MCB | −0.7 | −18.7 | 4.9 | |
| 1 + 2a→ TSB | 31.5 | −31.4 | 40.9 | |
| 1 + 2a→ 4a | −9.0 | −32.1 | 0.6 | |
| Nitromethane | 1 + 2a→ MCA | 0.1 | −11.2 | 3.4 |
| 1 + 2a→ TSA | 30.5 | −31.2 | 39.8 | |
| 1 + 2a→ 3a | −3.5 | −32.4 | 6.2 | |
| 1 + 2a→ MCB | −0.7 | −18.6 | 4.9 | |
| 1 + 2a→ TSB | 31.6 | −31.4 | 40.9 | |
| 1 + 2a→ 4a | −9.0 | −32.2 | 0.5 | |
| Water | 1 + 2a→ MCA | 0.1 | −11.2 | 3.4 |
| 1 + 2a→ TSA | 30.5 | −31.2 | 39.8 | |
| 1 + 2a→ 3a | −3.5 | −32.4 | 6.1 | |
| 1 + 2a→ MCB | −0.6 | −18.7 | 4.9 | |
| 1 + 2a→ TSB | 31.6 | −31.4 | 40.9 | |
| 1 + 2a→ 4a | −9.1 | −32.2 | 0.5 | |
| Toluene | 1 + 2b→ MCA | −0.6 | −15.0 | 3.9 |
| 1 + 2b→ TSA | 31.1 | −31.8 | 40.6 | |
| 1 + 2b→ 3b | −3.8 | −33.1 | 6.1 | |
| 1 + 2b→ MCB | −0.9 | −16.5 | 4.0 | |
| 1 + 2b→ TSB | 30.5 | −31.8 | 40.0 | |
| 1 + 2b→ 4b | −8.7 | −33.4 | 1.3 | |
| Toluene | 1 + 2c→ MCA | −0.4 | −15.5 | 4.2 |
| 1 + 2c→ TSA | 31.0 | −33.4 | 40.9 | |
| 1 + 2c→ 3c | −2.3 | −34.2 | 7.9 | |
| 1 + 2c→ MCB | −0.5 | 1169.2 | 4.8 | |
| 1 + 2c→ TSB | 32.8 | −33.5 | 42.8 | |
| 1 + 2c→ 4c | −9.7 | −33.8 | 0.4 | |
| Toluene | 1 + 2d→ MCA | −0.6 | −13.8 | 3.5 |
| 1 + 2d→ TSA | 30.8 | −30.6 | 40.0 | |
| 1 + 2d→ 3d | −2.5 | −32.2 | 7.1 | |
| 1 + 2d→ MCB | −0.6 | −16.3 | 4.3 | |
| 1 + 2d→ TSB | 33.4 | −30.8 | 42.6 | |
| 1 + 2d→ 4d | −6.8 | −32.1 | 2.8 | |
| Toluene | 1 + 2e→ MC | −0.3 | −20.5 | 5.8 |
| 1 + 2e→ TS | 31.1 | −35.3 | 41.7 | |
| 1 + 2e→ 3e | −4.2 | −35.4 | 6.3 |
| Solvent | Reaction | Structure | Interatomic Distances r (Å) | lC3C4 | lC5O1 | Δl | GEDT (e) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| O1-N2 | N2-N3 | N3-C4 | C4-C5 | C5-O1 | |||||||
| Toluene | 1 + 2a | 1 | 1.179 | 1.119 | |||||||
| 2a | 1.320 | ||||||||||
| MCA | 1.179 | 1.118 | 3.570 | 1.321 | 3.326 | ||||||
| TSA | 1.225 | 1.159 | 2.058 | 1.383 | 1.971 | 0.601 | 0.630 | 0.03 | 0.19 | ||
| 3a | 1.364 | 1.226 | 1.471 | 1.517 | 1.438 | ||||||
| MCB | 1.179 | 1.118 | 3.558 | 1.321 | 3.315 | ||||||
| TSB | 1.217 | 1.165 | 1.919 | 1.382 | 2.079 | 0.690 | 0.488 | 0.20 | 0.19 | ||
| 4a | 1.456 | 1.206 | 1.464 | 1.529 | 1.375 | ||||||
| Acetone | 1 + 2a | 1 | 1.179 | 1.118 | |||||||
| 2a | 1.321 | ||||||||||
| MCA | 1.178 | 1.118 | 3.407 | 1.321 | 3.294 | ||||||
| TSA | 1.224 | 1.158 | 2.068 | 1.383 | 1.970 | 0.594 | 0.633 | 0.04 | 0.18 | ||
| 3a | 1.361 | 1.227 | 1.471 | 1.516 | 1.441 | ||||||
| MCB | 1.179 | 1.118 | 3.612 | 1.321 | 3.308 | ||||||
| TSB | 1.217 | 1.165 | 1.913 | 1.382 | 2.091 | 0.693 | 0.478 | 0.21 | 0.17 | ||
| 4a | 1.458 | 1.206 | 1.464 | 1.528 | 1.375 | ||||||
| Nitromethane | 1 + 2a | 1 | 1.179 | 1.118 | |||||||
| 2a | 1.321 | ||||||||||
| MCA | 1.178 | 1.118 | 3.406 | 1.322 | 3.294 | ||||||
| TSA | 1.224 | 1.158 | 2.069 | 1.383 | 1.970 | 0.594 | 0.634 | 0.04 | 0.18 | ||
| 3a | 1.361 | 1.227 | 1.471 | 1.516 | 1.441 | ||||||
| MCB | 1.179 | 1.118 | 3.612 | 1.321 | 3.308 | ||||||
| TSB | 1.217 | 1.165 | 1.913 | 1.382 | 2.092 | 0.694 | 0.478 | 0.22 | 0.17 | ||
| 4a | 1.458 | 1.206 | 1.464 | 1.528 | 1.375 | ||||||
| Water | 1 + 2a | 1 | 1.179 | 1.118 | |||||||
| 2a | 1.321 | ||||||||||
| MCA | 1.178 | 1.118 | 3.398 | 1.322 | 3.296 | ||||||
| TSA | 1.224 | 1.158 | 2.069 | 1.383 | 1.970 | 0.594 | 0.634 | 0.04 | 0.18 | ||
| 3a | 1.360 | 1.227 | 1.471 | 1.516 | 1.442 | ||||||
| MCB | 1.179 | 1.118 | 3.612 | 1.321 | 3.309 | ||||||
| TSB | 1.217 | 1.165 | 1.913 | 1.382 | 2.093 | 0.694 | 0.477 | 0.22 | 0.17 | ||
| 4a | 1.459 | 1.206 | 1.464 | 1.528 | 1.375 | ||||||
| Toluene | 1 + 2b | 2b | 1.325 | ||||||||
| MCA | 1.178 | 1.119 | 3.305 | 1.325 | 3.329 | ||||||
| TSA | 1.225 | 1.158 | 2.071 | 1.388 | 1.985 | 0.598 | 0.617 | 0.02 | 0.21 | ||
| 3b | 1.369 | 1.223 | 1.477 | 1.526 | 1.435 | ||||||
| MCB | 1.179 | 1.118 | 3.512 | 1.325 | 3.153 | ||||||
| TSB | 1.216 | 1.166 | 1.926 | 1.386 | 2.122 | 0.683 | 0.466 | 0.22 | 0.19 | ||
| 4b | 1.442 | 1.211 | 1.463 | 1.527 | 1.383 | ||||||
| Toluene | 1 + 2c | 2c | 1.317 | ||||||||
| MCA | 1.181 | 1.118 | 3.981 | 1.317 | 3.150 | ||||||
| TSA | 1.227 | 1.152 | 2.098 | 1.389 | 1.886 | 0.556 | 0.686 | 0.13 | 0.34 | ||
| 3c | 1.353 | 1.232 | 1.452 | 1.512 | 1.436 | ||||||
| MCB | 1.179 | 1.118 | 3.675 | 1.317 | 3.113 | ||||||
| TSB | 1.212 | 1.166 | 1.866 | 1.386 | 2.057 | 0.714 | 0.463 | 0.25 | 0.34 | ||
| 4c | 1.528 | 1.193 | 1.451 | 1.536 | 1.338 | ||||||
| Toluene | 1 + 2d | 2d | 1.326 | ||||||||
| MCA | 1.180 | 1.118 | 3.468 | 1.327 | 3.174 | ||||||
| TSA | 1.224 | 1.161 | 2.041 | 1.389 | 2.015 | 0.607 | 0.608 | 0.00 | 0.20 | ||
| 3d | 1.361 | 1.228 | 1.465 | 1.525 | 1.447 | ||||||
| MCB | 1.178 | 1.119 | 3.463 | 1.326 | 3.362 | ||||||
| TSB | 1.220 | 1.166 | 1.928 | 1.388 | 2.070 | 0.690 | 0.491 | 0.20 | 0.19 | ||
| 4d | 1.460 | 1.206 | 1.472 | 1.536 | 1.371 | ||||||
| Toluene | 1 + 2e | 2e | 1.318 | ||||||||
| MC | 1.180 | 1.118 | 3.493 | 1.319 | 3.038 | ||||||
| TS | 1.221 | 1.160 | 1.982 | 1.386 | 1.968 | 0.647 | 0.574 | 0.07 | 0.37 | ||
| 3e | 1.412 | 1.217 | 1.465 | 1.523 | 1.380 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Regioselectivity and the Molecular Mechanism of [3 + 2] Cycloaddition Reactions between Nitrous Oxide and Conjugated Nitroalkenes: A DFT Computational Study. Molecules 2022, 27, 8441. https://doi.org/10.3390/molecules27238441
Dresler E, Wróblewska A, Jasiński R. Understanding the Regioselectivity and the Molecular Mechanism of [3 + 2] Cycloaddition Reactions between Nitrous Oxide and Conjugated Nitroalkenes: A DFT Computational Study. Molecules. 2022; 27(23):8441. https://doi.org/10.3390/molecules27238441
Chicago/Turabian StyleDresler, Ewa, Aneta Wróblewska, and Radomir Jasiński. 2022. "Understanding the Regioselectivity and the Molecular Mechanism of [3 + 2] Cycloaddition Reactions between Nitrous Oxide and Conjugated Nitroalkenes: A DFT Computational Study" Molecules 27, no. 23: 8441. https://doi.org/10.3390/molecules27238441