
Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence



Abstract
:1. Introduction
2. Pharmacology and Mechanism of Action of PDEs and Ibudilast in the CNS
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Amyotrophic Lateral Sclerosis
2.4. Multiple Sclerosis
2.5. Other Neurodegenerative Diseases
3. Challenges and Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calderon-Garciduenas, L. Common Fatal Neurodegenerative Diseases Revisited: Beyond Age, Comorbidities, and Devastating Terminal Neuropathology There Is Hope with Prevention. Front. Neurol. 2022, 13, 901447. [Google Scholar] [CrossRef]
- Chaudhuri, A. Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 2013, 120, 1463–1466. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. APOE Genotype and Alzheimer’s Disease: The Influence of Lifestyle and Environmental Factors. ACS Chem. Neurosci. 2021, 12, 2749–2764. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Bozi, M.; Simitsi, A.M.; Koros, C.; Antonelou, R.; Papagiannakis, N.; Maniati, M.; Poula, D.; Stamelou, M.; Vassilatis, D.K.; et al. The relationship between environmental factors and different Parkinson’s disease subtypes in Greece: Data analysis of the Hellenic Biobank of Parkinson’s disease. Park. Relat. Disord. 2019, 67, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Shaikh, M.F.; Piperi, C. Fractalkine (CX3CL1) signaling and neuroinflammation in Parkinson’s disease: Potential clinical and therapeutic implications. Pharmacol. Res. 2020, 158, 104930. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [Green Version]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Piperi, C. Exploring the role of high-mobility group box 1 (HMGB1) protein in the pathogenesis of Huntington’s disease. J. Mol. Med. 2020, 98, 325–334. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Julian, T.; Shaikh, M.F.; Piperi, C. Pivotal Role of Fyn Kinase in Parkinson’s Disease and Levodopa-Induced Dyskinesia: A Novel Therapeutic Target? Mol. Neurobiol. 2021, 58, 1372–1391. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. JAD 2010, 20 (Suppl. 2), S369–S379. [Google Scholar] [CrossRef] [Green Version]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal dysfunction in neurodegeneration: Emerging concepts and methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef]
- Nasiri, E.; Sankowski, R.; Dietrich, H.; Oikonomidi, A.; Huerta, P.T.; Popp, J.; Al-Abed, Y.; Bacher, M. Key role of MIF-related neuroinflammation in neurodegeneration and cognitive impairment in Alzheimer’s disease. Mol. Med. 2020, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life 2012, 64, 965–970. [Google Scholar] [CrossRef]
- Zhong, J.; Xie, J.; Xiao, J.; Li, D.; Xu, B.; Wang, X.; Wen, H.; Zhou, Z.; Cheng, Y.; Xu, J.; et al. Inhibition of PDE4 by FCPR16 induces AMPK-dependent autophagy and confers neuroprotection in SH-SY5Y cells and neurons exposed to MPP(+)-induced oxidative insult. Free Radic. Biol. Med. 2019, 135, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Coffey, C.S.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Conwit, R.; Naismith, R.; et al. Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp. Clin. Trials 2016, 50, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Gibson, L.C.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; Mackenzie, F.L.; Nagasawa, M.; Stevens, P.A.; Mackenzie, S.J. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur. J. Pharmacol. 2006, 538, 39–42. [Google Scholar] [CrossRef]
- Wu, N.C.; Wang, J.J. Ibudilast, a Phosphodiesterase Inhibitor and Toll-Like Receptor-4 Antagonist, Improves Hemorrhagic Shock and Reperfusion-Induced Left Ventricular Dysfunction by Reducing Myocardial Tumor Necrosis Factor alpha. Transplant. Proc. 2020, 52, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Suzumura, A.; Ito, A.; Yoshikawa, M.; Sawada, M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999, 837, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, Y.; Nakamura, Y.; Tsuji, K.; Shibata, T.; Kataoka, K. Ibudilast protects against neuronal damage induced by glutamate in cultured hippocampal neurons. Clin. Exp. Pharmacol. Physiol. 1996, 23, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Lin, J.X.; Ihara, M.; Ohtani, R.; Shibata, M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Res. 2003, 992, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Tamano, R.; Yoneda, S.; Omachi, S.; Yogo, E.; Rokushima, M.; Shinohara, S.; Sakaguchi, G.; Hasegawa, M.; Asaki, T. Ibudilast produces anti-allodynic effects at the persistent phase of peripheral or central neuropathic pain in rats: Different inhibitory mechanism on spinal microglia from minocycline and propentofylline. Eur. J. Pharmacol. 2018, 833, 263–274. [Google Scholar] [CrossRef]
- Ledeboer, A.; Hutchinson, M.R.; Watkins, L.R.; Johnson, K.W. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin. Investig. Drugs 2007, 16, 935–950. [Google Scholar] [CrossRef]
- Kiebala, M.; Maggirwar, S.B. Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells. PLoS ONE 2011, 6, e18633. [Google Scholar] [CrossRef] [Green Version]
- Yagi, K.; Tada, Y.; Kitazato, K.T.; Tamura, T.; Satomi, J.; Nagahiro, S. Ibudilast inhibits cerebral aneurysms by down-regulating inflammation-related molecules in the vascular wall of rats. Neurosurgery 2010, 66, 551–559, discussion 559. [Google Scholar] [CrossRef]
- Yoshioka, M.; Suda, N.; Mori, K.; Ueno, K.; Itoh, Y.; Togashi, H.; Matsumoto, M. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacol. Res. 2002, 45, 305–311. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cho, E.; Ko, Y.E.; Kim, I.; Lee, K.J.; Kwon, S.U.; Kang, D.W.; Kim, J.S. Ibudilast, a phosphodiesterase inhibitor with anti-inflammatory activity, protects against ischemic brain injury in rats. Brain Res. 2012, 1431, 97–106. [Google Scholar] [CrossRef]
- Johnston, I.N.; Tan, M.; Cao, J.; Matsos, A.; Forrest, D.R.L.; Si, E.; Fardell, J.E.; Hutchinson, M.R. Ibudilast reduces oxaliplatin-induced tactile allodynia and cognitive impairments in rats. Behav. Brain Res. 2017, 334, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Matsukane, R.; Egashira, N.; Tsuchiya, Y.; Fu, R.; Yamamoto, S.; Hirota, T.; Ieiri, I. Neuroprotective effects of ibudilast against tacrolimus induced neurotoxicity. Toxicol. Appl. Pharmacol. 2022, 449, 116112. [Google Scholar] [CrossRef] [PubMed]
- Poland, R.S.; Hahn, Y.; Knapp, P.E.; Beardsley, P.M.; Bowers, M.S. Ibudilast attenuates expression of behavioral sensitization to cocaine in male and female rats. Neuropharmacology 2016, 109, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Mei, Z.; Zhong, K.L.; Hu, M.; Long, Y.; Miao, M.X.; Li, N.; Yan, T.H.; Hong, H. Pretreatment with antiasthmatic drug ibudilast ameliorates Abeta 1-42-induced memory impairment and neurotoxicity in mice. Pharmacol. Biochem. Behav. 2014, 124, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Maragakis, N.; Bedlack, R.S.; Goyal, N.; Meyer, J.A.; Genge, A.; Bodkin, C.; Maiser, S.; Staff, N.; Zinman, L.; et al. MN-166 (ibudilast) in amyotrophic lateral sclerosis in a Phase IIb/III study: COMBAT-ALS study design. Neurodegener. Dis. Manag. 2021, 11, 431–443. [Google Scholar] [CrossRef]
- Naismith, R.T.; Bermel, R.A.; Coffey, C.S.; Goodman, A.D.; Fedler, J.; Kearney, M.; Klawiter, E.C.; Nakamura, K.; Narayanan, S.; Goebel, C.; et al. Effects of Ibudilast on MRI Measures in the Phase 2 SPRINT-MS Study. Neurology 2021, 96, e491–e500. [Google Scholar] [CrossRef]
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin. Pharmacother. 2009, 10, 2897–2904. [Google Scholar] [CrossRef]
- Crocetti, L.; Floresta, G.; Cilibrizzi, A.; Giovannoni, M.P. An Overview of PDE4 Inhibitors in Clinical Trials: 2010 to Early 2022. Molecules 2022, 27, 4964. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Perez-Torres, S.; Cortes, R.; Tolnay, M.; Probst, A.; Palacios, J.M.; Mengod, G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp. Neurol. 2003, 182, 322–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kumar, K.; Deshmukh, R.; Sharma, P.L. Phosphodiesterases: Regulators of cyclic nucleotide signals and novel molecular target for movement disorders. Eur. J. Pharmacol. 2013, 714, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Kurotani, T.; Komatsu, Y.; Kawanokuchi, J.; Kato, H.; Mitsuma, N.; Suzumura, A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 2004, 46, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Ohta, S.; Kasuya, N.; Sakita, S.; Ashikaga, T.; Isobe, M. Ibudilast: A non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovasc. Drug Rev. 2001, 19, 215–225. [Google Scholar] [CrossRef]
- Souness, J.E.; Villamil, M.E.; Scott, L.C.; Tomkinson, A.; Giembycz, M.A.; Raeburn, D. Possible role of cyclic AMP phosphodiesterases in the actions of ibudilast on eosinophil thromboxane generation and airways smooth muscle tone. Br. J. Pharmacol. 1994, 111, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolan, P.; Gibbons, J.A.; He, L.; Chang, E.; Jones, D.; Gross, M.I.; Davidson, J.B.; Sanftner, L.M.; Johnson, K.W. Ibudilast in healthy volunteers: Safety, tolerability and pharmacokinetics with single and multiple doses. Br. J. Clin. Pharmacol. 2008, 66, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Sanftner, L.M.; Gibbons, J.A.; Gross, M.I.; Suzuki, B.M.; Gaeta, F.C.; Johnson, K.W. Cross-species comparisons of the pharmacokinetics of ibudilast. Xenobiotica Fate Foreign Compd. Biol. Syst. 2009, 39, 964–977. [Google Scholar] [CrossRef]
- Babu, S.; Hightower, B.G.; Chan, J.; Zurcher, N.R.; Kivisakk, P.; Tseng, C.J.; Sanders, D.L.; Robichaud, A.; Banno, H.; Evora, A.; et al. Ibudilast (MN-166) in amyotrophic lateral sclerosis- an open label, safety and pharmacodynamic trial. NeuroImage Clin. 2021, 30, 102672. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impey, S.; Mark, M.; Villacres, E.C.; Poser, S.; Chavkin, C.; Storm, D.R. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 1996, 16, 973–982. [Google Scholar] [CrossRef]
- Su, Y.; Ryder, J.; Ni, B. Inhibition of Abeta production and APP maturation by a specific PKA inhibitor. FEBS Lett. 2003, 546, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yang, X.M.; Zhuo, Y.Y.; Zhou, H.; Lin, H.B.; Cheng, Y.F.; Xu, J.P.; Zhang, H.T. The phosphodiesterase-4 inhibitor rolipram reverses Abeta-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int. J. Neuropsychopharmacol. 2012, 15, 749–766. [Google Scholar] [CrossRef] [Green Version]
- Maki, T.; Okamoto, Y.; Carare, R.O.; Hase, Y.; Hattori, Y.; Hawkes, C.A.; Saito, S.; Yamamoto, Y.; Terasaki, Y.; Ishibashi-Ueda, H.; et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular beta-amyloid. Ann. Clin. Transl. Neurol. 2014, 1, 519–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.Y.; Yang, M.X.; Zhang, Y.H.; Zheng, V.; Zhang, H.T.; Gurney, M.E.; Xu, Y.; O’Donnell, J.M. Protection from Amyloid beta Peptide-Induced Memory, Biochemical, and Morphological Deficits by a Phosphodiesterase-4D Allosteric Inhibitor. J. Pharmacol. Exp. Ther. 2019, 371, 250–259. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Takiguchi, O.; Nishiyama, A.; Mori, H. Cilostazol prevents amyloid beta peptide(25-35)-induced memory impairment and oxidative stress in mice. Br. J. Pharmacol. 2010, 161, 1899–1912. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Kim, J.H.; Bae, S.S.; Hong, K.W.; Lee, D.S.; Leem, J.Y.; Choi, B.T.; Shin, H.K. Protective effect of the phosphodiesterase III inhibitor cilostazol on amyloid beta-induced cognitive deficits associated with decreased amyloid beta accumulation. Biochem. Biophys. Res. Commun. 2011, 408, 602–608. [Google Scholar] [CrossRef]
- Schaler, A.W.; Myeku, N. Cilostazol, a phosphodiesterase 3 inhibitor, activates proteasome-mediated proteolysis and attenuates tauopathy and cognitive decline. Transl. Res. J. Lab. Clin. Med. 2018, 193, 31–41. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Bae, S.S.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol Upregulates Autophagy via SIRT1 Activation: Reducing Amyloid-beta Peptide and APP-CTFbeta Levels in Neuronal Cells. PLoS ONE 2015, 10, e0134486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prickaerts, J.; Heckman, P.R.A.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Lee, H.; Yoo, H.B.; Choi, J.S.; Jung, H.Y.; Yoon, E.J.; Kim, H.; Jung, Y.H.; Lee, H.Y.; Kim, Y.K. Efficacy of Cilostazol Administration in Alzheimer’s Disease Patients with White Matter Lesions: A Positron-Emission Tomography Study. Neurother. J. Am. Soc. Exp. NeuroTher. 2019, 16, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, G.; Wallace, C.H.; Chaudry, O.; Liu, Q.; Qiu, Y.; Xie, L.; Rockwell, P.; Figueiredo-Pereira, M.E.; Serrano, P.A. Repurposing ibudilast to mitigate Alzheimer’s disease by targeting inflammation. Brain A J. Neurol. 2022, Online ahead of print. [Google Scholar] [CrossRef]
- Schwenkgrub, J.; Zaremba, M.; Joniec-Maciejak, I.; Cudna, A.; Mirowska-Guzel, D.; Kurkowska-Jastrzebska, I. The phosphodiesterase inhibitor, ibudilast, attenuates neuroinflammation in the MPTP model of Parkinson’s disease. PLoS ONE 2017, 12, e0182019. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; Melchiorri, G.; Nuccetelli, V.; D’Angelo, V.; Martorana, A.; Sorge, R.; Castelli, V.; Bernardi, G.; Sancesario, G. PDE10A and PDE10A-dependent cAMP catabolism are dysregulated oppositely in striatum and nucleus accumbens after lesion of midbrain dopamine neurons in rat: A key step in parkinsonism physiopathology. Neurobiol. Dis. 2011, 43, 293–303. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Lorenzo, B.J.; Beal, M.F. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp. Neurol. 2008, 211, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Yu, H.; Huang, C.; Zhong, Q.; Chen, Y.; Xie, J.; Zhou, Z.; Xu, J.; Wang, H. Inhibition of phosphodiesterase 4 by FCPR16 protects SH-SY5Y cells against MPP(+)-induced decline of mitochondrial membrane potential and oxidative stress. Redox Biol. 2018, 16, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; D’Angelo, V.; Esposito, Z.; Nuccetelli, V.; Sorge, R.; Martorana, A.; Stefani, A.; Bernardi, G.; Sancesario, G. Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: New aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 2008, 28, 941–950. [Google Scholar] [CrossRef]
- Corcia, P.; Beltran, S.; Bakkouche, S.E.; Couratier, P. Therapeutic news in ALS. Rev. Neurol. 2021, 177, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Chattopadhyay, M.; Whitelegge, J.; Valentine, J.S. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr. Top. Med. Chem. 2012, 12, 2560–2572. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain J. Neurol. 2019, 142, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Bicchi, I.; Morena, F.; Argentati, C.; Nodari, L.R.; Emiliani, C.; Gelati, M.; Vescovi, A.L.; Martino, S. Storage of Mutant Human SOD1 in Non-Neural Cells from the Type-1 Amyotrophic Lateral Sclerosis rat(G93A) Model Correlated with the Lysosomes’ Dysfunction. Biomedicines 2021, 9, 1080. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Implication of HMGB1 signaling pathways in Amyotrophic lateral sclerosis (ALS): From molecular mechanisms to pre-clinical results. Pharmacol. Res. 2020, 156, 104792. [Google Scholar] [CrossRef]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.; Trojanowski, J.Q. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [Green Version]
- Downer, O.M.; Marcus, R.E.G.; Zurcher, N.R.; Hooker, J.M. Tracing the History of the Human Translocator Protein to Recent Neurodegenerative and Psychiatric Imaging. ACS Chem. Neurosci. 2020, 11, 2192–2200. [Google Scholar] [CrossRef]
- Alshikho, M.J.; Zurcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C.; et al. Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: The new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem. Biophys. Res. Commun. 2020, 526, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bravver, E.; Sanjak, M.; Langford, V.; Moore, L.; Smith, N.; Lucas, N.; Nichols, M.; Lary, C.; Newell-Sturdivant, A.; et al. Adaptive Design Single Center Phosphodiesterase Type 4 (PDE4) Inhibitor-Ibudilast (MN-166-ALS-1201) Phase 1b/2a Clinical Trial Double-Blind (DB) with Open Label Extension (OLE) [ NCT02238626 ] for Amyotrophic Lateral Sclerosis (ALS ) Patients [ 1 ] Not Requiring Non-Invasive Ventilation (no NIV) up to 5 years (Early Cohort-EC) and [ 2 ] Requiring Non-Invasive Ventilation (NIV) up to 10 years (Advanced NIV Cohort-ANC) from Disease Onset-Report of Clinical Trial DB, OLE and Post-Treatment Cessation Epochs-Per-Protocol (PP) Treatment Completion Associated with Improved Survival and Post Treatment Cessation Loss of Muscle Strength (P3.127). Neurology 2017, 88, P3.127. [Google Scholar]
- Loma, I.; Heyman, R. Multiple sclerosis: Pathogenesis and treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef]
- Groppa, S.; Gonzalez-Escamilla, G.; Eshaghi, A.; Meuth, S.G.; Ciccarelli, O. Linking immune-mediated damage to neurodegeneration in multiple sclerosis: Could network-based MRI help? Brain Commun. 2021, 3, fcab237. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Fransen, N.L.; Touil, H.; Michailidou, I.; Huitinga, I.; Gommerman, J.L.; Bar-Or, A.; Ramaglia, V. Accumulation of meningeal lymphocytes correlates with white matter lesion activity in progressive multiple sclerosis. JCI Insight 2022, 7, e151683. [Google Scholar] [CrossRef]
- Hagman, S.; Raunio, M.; Rossi, M.; Dastidar, P.; Elovaara, I. Disease-associated inflammatory biomarker profiles in blood in different subtypes of multiple sclerosis: Prospective clinical and MRI follow-up study. J. Neuroimmunol. 2011, 234, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Covacu, R.; Sunnemark, D.; Danilov, A.I.; Dal Bianco, A.; Khademi, M.; Wallstrom, E.; Lobell, A.; Brundin, L.; Lassmann, H.; et al. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 2008, 84, 1248–1255. [Google Scholar] [CrossRef] [Green Version]
- Paudel, Y.N.; Angelopoulou, E.; Bhuvan, K.C.; Piperi, C.; Othman, I. High mobility group box 1 (HMGB1) protein in Multiple Sclerosis (MS): Mechanisms and therapeutic potential. Life Sci. 2019, 238, 116924. [Google Scholar] [CrossRef]
- Sommer, N.; Loschmann, P.A.; Northoff, G.H.; Weller, M.; Steinbrecher, A.; Steinbach, J.P.; Lichtenfels, R.; Meyermann, R.; Riethmuller, A.; Fontana, A.; et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat. Med. 1995, 1, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Genain, C.P.; Roberts, T.; Davis, R.L.; Nguyen, M.H.; Uccelli, A.; Faulds, D.; Li, Y.; Hedgpeth, J.; Hauser, S.L. Prevention of autoimmune demyelination in non-human primates by a cAMP-specific phosphodiesterase inhibitor. Proc. Natl. Acad. Sci. USA 1995, 92, 3601–3605. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Zielasek, J.; Kollner, G.; Donhauser, T.; Toyka, K.; Hartung, H.P. Preventive but not therapeutic application of Rolipram ameliorates experimental autoimmune encephalomyelitis in Lewis rats. J. Neuroimmunol. 1996, 68, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Richert, N.; Howard, T.; Packer, A.N.; Blevins, G.; Ohayon, J.; McFarland, H.F.; Sturzebecher, C.S.; Martin, R. Treatment with the phosphodiesterase type-4 inhibitor rolipram fails to inhibit blood--brain barrier disruption in multiple sclerosis. Mult. Scler. 2009, 15, 1206–1214. [Google Scholar] [CrossRef]
- Fujimoto, T.; Sakoda, S.; Fujimura, H.; Yanagihara, T. Ibudilast, a phosphodiesterase inhibitor, ameliorates experimental autoimmune encephalomyelitis in Dark August rats. J. Neuroimmunol. 1999, 95, 35–42. [Google Scholar] [CrossRef]
- Barkhof, F.; Hulst, H.E.; Drulovic, J.; Uitdehaag, B.M.; Matsuda, K.; Landin, R.; Investigators, M.N. Ibudilast in relapsing-remitting multiple sclerosis: A neuroprotectant? Neurology 2010, 74, 1033–1040. [Google Scholar] [CrossRef]
- Fox, R.J.; Raska, P.; Barro, C.; Karafa, M.; Konig, V.; Bermel, R.A.; Chase, M.; Coffey, C.S.; Goodman, A.D.; Klawiter, E.C.; et al. Neurofilament light chain in a phase 2 clinical trial of ibudilast in progressive multiple sclerosis. Mult. Scler. 2021, 27, 2014–2022. [Google Scholar] [CrossRef]
- Bermel, R.A.; Fedler, J.K.; Kaiser, P.; Novalis, C.; Schneebaum, J.; Klingner, E.A.; Williams, D.; Yankey, J.W.; Ecklund, D.J.; Chase, M.; et al. Optical coherence tomography outcomes from SPRINT-MS, a multicenter, randomized, double-blind trial of ibudilast in progressive multiple sclerosis. Mult. Scler. 2021, 27, 1384–1390. [Google Scholar] [CrossRef]
- Goodman, A.D.; Fedler, J.K.; Yankey, J.; Klingner, E.A.; Ecklund, D.J.; Goebel, C.V.; Bermel, R.A.; Chase, M.; Coffey, C.S.; Klawiter, E.C.; et al. Response to ibudilast treatment according to progressive multiple sclerosis disease phenotype. Ann. Clin. Transl. Neurol. 2021, 8, 111–118. [Google Scholar] [CrossRef]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; De Angelis, F.; van de Pavert, S.H.; Cawley, N.; De Stefano, N.; et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampa, C.; Laurenti, D.; Anzilotti, S.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS ONE 2010, 5, e13417. [Google Scholar] [CrossRef] [PubMed]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diabetes Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.D.; Fischer, T.T.; Abreu, D.; Arroyo, A.; Urano, F.; Ehrlich, B.E. Calpain inhibitor and ibudilast rescue beta cell functions in a cellular model of Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 17389–17398. [Google Scholar] [CrossRef] [PubMed]
- Benbow, J.H.; Mann, T.; Keeler, C.; Fan, C.; Hodsdon, M.E.; Lolis, E.; DeGray, B.; Ehrlich, B.E. Inhibition of paclitaxel-induced decreases in calcium signaling. J. Biol. Chem. 2012, 287, 37907–37916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, L.R., Jr.; Harbeck, M.; Kaihara, K.; Chepurny, O.; Kitiphongspattana, K.; Graf, O.; Nikolaev, V.O.; Lohse, M.J.; Holz, G.G.; Roe, M.W. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J. Biol. Chem. 2005, 280, 31294–31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Yucel, Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007, 18, 110–114. [Google Scholar] [CrossRef]
- Cueva Vargas, J.L.; Belforte, N.; Di Polo, A. The glial cell modulator ibudilast attenuates neuroinflammation and enhances retinal ganglion cell viability in glaucoma through protein kinase A signaling. Neurobiol. Dis. 2016, 93, 156–171. [Google Scholar] [CrossRef]
- Bollen, E.; Puzzo, D.; Rutten, K.; Privitera, L.; De Vry, J.; Vanmierlo, T.; Kenis, G.; Palmeri, A.; D’Hooge, R.; Balschun, D.; et al. Improved long-term memory via enhancing cGMP-PKG signaling requires cAMP-PKA signaling. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2014, 39, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Rile, G.; Yatomi, Y.; Qi, R.; Satoh, K.; Ozaki, Y. Potentiation of ibudilast inhibition of platelet aggregation in the presence of endothelial cells. Thromb. Res. 2001, 102, 239–246. [Google Scholar] [CrossRef]
- Corsi, M.M.; Licastro, F.; Porcellini, E.; Dogliotti, G.; Galliera, E.; Lamont, J.L.; Innocenzi, P.J.; Fitzgerald, S.P. Reduced plasma levels of P-selectin and L-selectin in a pilot study from Alzheimer disease: Relationship with neurodegeneration. Biogerontology 2011, 12, 451–454. [Google Scholar] [CrossRef]
- Drake, J.D.; Chambers, A.B.; Ott, B.R.; Daiello, L.A.; Alzheimer’s Disease Neuroimaging, I. Peripheral Markers of Vascular Endothelial Dysfunction Show Independent but Additive Relationships with Brain-Based Biomarkers in Association with Functional Impairment in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2021, 80, 1553–1565. [Google Scholar] [CrossRef]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Sadighi Akha, A.A. Aging and the immune system: An overview. J. Immunol. Methods 2018, 463, 21–26. [Google Scholar] [CrossRef] [PubMed]
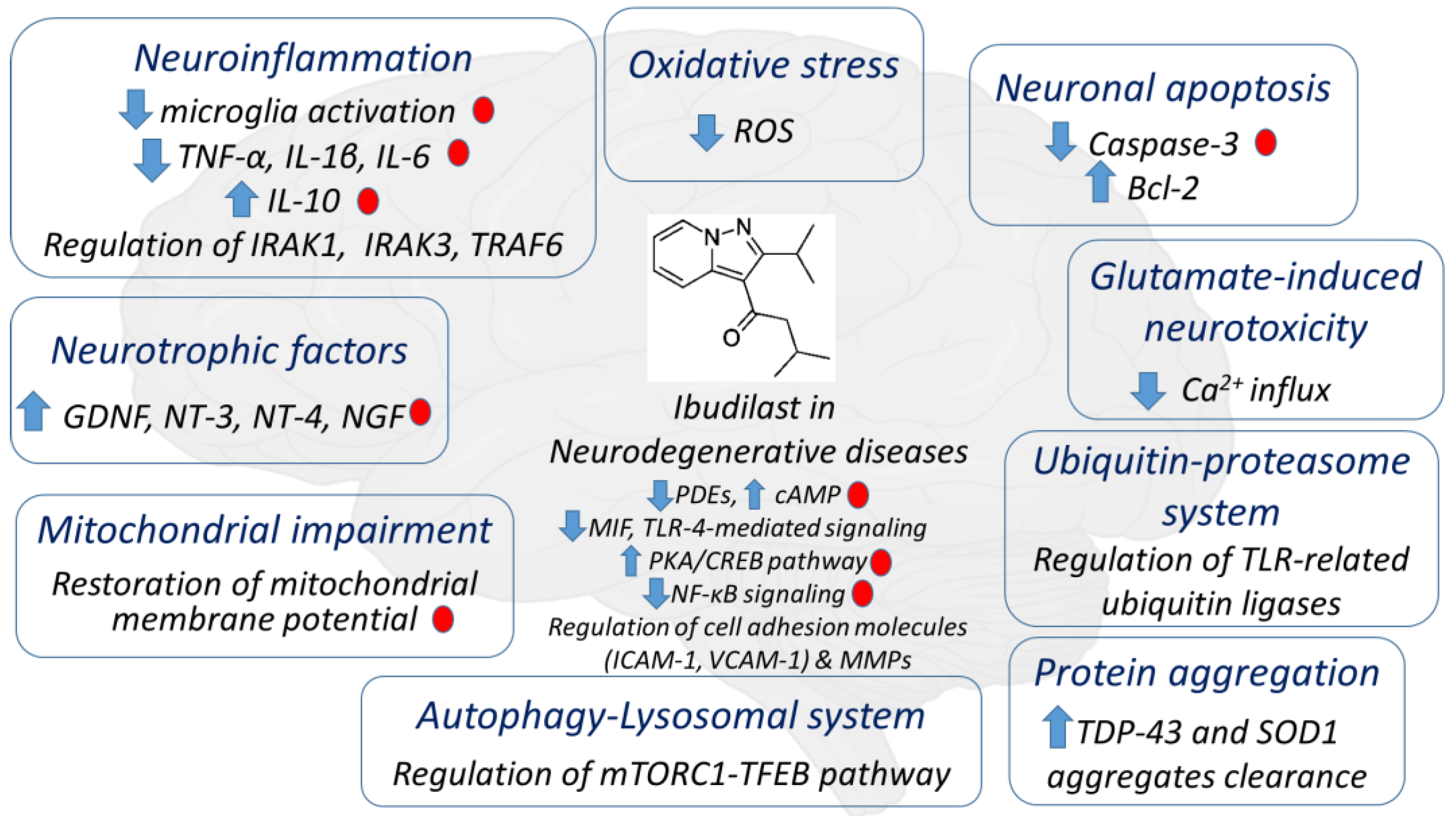

{kind=link}
| PDE Type | Distribution | Inhibitors | Reference |
|---|---|---|---|
| PDE1 | Heart, lungs, brain, smooth muscle. | Ibudilast, nimodipine, dioclein, IC86340, IC224, IC295. | [22,40] |
| PDE2 | Heart, kidneys, brain, platelets, adrenal glands, lungs, liver, endothelial cells. | Ibudilast, oxindole, EHNA, ND7001, BAY-60–7750, PDP, IC933. | [22,40,45] |
| PDE3 | Heart, kidneys, brain, lungs, smooth muscle, liver, platelets, adipocytes, immune cells. | Ibudilast, cilostazol, milrinone, cilostamide, siguazodan. | [22,40,56,58,59] |
| PDE4 | Heart, kidneys, brain, platelets, Sertolli cells, liver, smooth muscle, lungs, endothelial cells, immune cells. | Ibudilast, rolipram, cilomast, roflumilast, NCS 613. | [22,40,45,55,57] |
| PDE5 | Platelets, heart, lungs, smooth muscle, brain, endothelial cells. | Ibudilast, DMPPO, zaprinast, vardenafil, sildenafil, tadalafil | [40,45] |
| PDE6 | Lungs, pineal gland, photoreceptors. | DMPPO, zaprinast, sildenafil, vardenafil. | [40] |
| PDE7 | Heart, skeletal muscle, T lymphocytes, kidneys, brain, pancreas. | IC242, BRL 50481, ASB16165. | [40] |
| PDE8 | Brain, eyes, testes, liver, heart, skeletal muscle, kidneys, thyroid, ovaries, T lymphocytes. | PF-04957325. | [40,42] |
| PDE9 | Lungs, kidneys, liver, brain. | PF-04447943, BAY-73–6691 | [40] |
| PDE10 | Brain, testes, thyroid. | Ibudilast, MP-10, Papaverine, TP-10. | [20,40,66,67] |
| PDE11 | Heart, liver, skeletal muscle, pituitary gland, prostate. | Ibudilast, non-selective. | [20,40] |
| PDE Inhibitor | PDE Target | Clinical Trials | Main Effects and Mechanism of Action in Neurodegenerative Diseases | Reference |
|---|---|---|---|---|
| Alzheimer’s Disease | ||||
| Rolipram | PDE4 | - | Inhibition of Aβ-mediated cognitive decline, via the regulation of neuroinflammatory and apoptotic responses in rats through cAMP/CREB signaling. | [55] |
| Zatomilast | PDE4 | Phase 1 clinical trials (NCT02648672, NCT02840279, NCT03030105); Phase 2 clinical trial (NCT03817684). | Improvement of memory, prevention of the loss of dendrites and spine density, inhibition of amyloid-beta-induced reduction of CREB, BDNF and NGF in the hippocampus of mice models of AD. | [57] |
| Cilostazol | PDE3 | Randomized, placebo-controlled phase 4 clinical trial (NCT01409564). | Prevention of amyloid-beta-induced oxidative stress and memory impairment. | [58] |
| - | Prevention of APOE-mediated amyloid-beta aggregation in mice. | [59] | ||
| - | Induction of proteasome-mediated proteolysis, suppression of tauopathy and attenuation of cognitive impairment. | [60] | ||
| - | Regulation of autophagy by upregulating SIRT1, and enhancement of amyloid-beta clearance and cell viability. | [61] | ||
| Parkinson’s disease | ||||
| Rolipram | PDE4 | - | Inhibition of MPTP-induced dopamine loss in the striatum of mice, and prevention of dopaminergic neuronal loss in the SN. | [68] |
| FCPR16 | PDE4 | - | Prevention of the MPP+-induced reduction of oxidative stress and the potential of the mitochondrial membrane. | [69] |
| Zaprinast | PDE6, 5, 11 and 9 | - | Prevention of cAMP and cGMP dysregulation in levodopa-induced dyskinesias in 6-OHDA-treated rat models of PD. | [70] |
| Multiple Sclerosis | ||||
| Rolipram | PDE4 | Phase 2 clinical trial (NCT00011375). | Prevention of the clinical signs of demyelination in EAE rat models, reduction of TNF-α production in MBP-specific T cells. | [89] |
| - | Reduction of TNF-α levels, prevention of clinical signs of MS and neuroimaging abnormalities on MRI in marmoset models of EAE. | [90] | ||
| Neurodegenerative Disease | Type of Study | Model | Main Findings | Reference |
|---|---|---|---|---|
| Alzheimer’s disease | In vitro | Cultured hippocampal neurons from rats. | -Ibudilast could protect against glutamate-induced neurotoxicity and increase intracellular cAMP levels. -Ibudilast treatment was associated with reduced glutamate induced Ca2+ influx. | [23] |
| In vivo | Sprague Dawley rats rat models | -Ibudilast could reverse the LPS- and INF-γ-induced inhibition of LTP in the CA1 region of hippocampus. | [44] | |
| In vivo | Amyloid-beta-injected mice mouse models of AD. | -Ibudilast pretreatment could prevent amyloid-beta-induced memory, spatial learning impairment, and neurotoxicity. -Ibudilast could inhibit the production of pro-inflammatory cytokines NF-κB p65 and TNF-α, prevent the activation of the pro-apoptotic protein caspase-3, and suppress the downregulation of the anti-apoptotic protein Bcl-2 in the cortex and hippocampus. | [34] | |
| In vivo | Fisher transgenic 344-AD rats. | -Long-term ibudilast treatment was associated with lower hippocampal-dependent spatial memory impairment, hippocampal amyloid-beta plaque deposition, tau paired-helical filament burden, and microgliosis. -RNA sequencing of hippocampal samples showed that ibudilast could affect the expression of the TLR, as well as the ubiquitin–proteasome pathways. -Ibudilast could downregulate the activity of IRAK1 by elevating the expression of IRAK3, affecting the levels of TRAF6 and possibly other TLR-related ubiquitin ligase. | [65] | |
| Parkinson’s disease | In vivo | MPTP mouse models of PD. | -Pretreatment with ibudilast was associated with reduced astroglia activity and increased GDNF in the striatum. -Ibudilast could also suppress the production of pro-inflammatory cytokines, including IL-6, IL-1β and TNF-α. -Ibudilast did not alter the dopaminergic neuronal cell survival and TH levels in the striatum seven days after the acute MPTP insult in this study. | [66] |
| Amyotrophic Lateral Sclerosis | In vitro | HEK293 and NSC-34 cells. | -Ibudilast treatment could stimulate the clearance of SOD1 and TDP-43 aggregates, via induction of autophagy, increase in autolysosomes, and enhancement of lysosomal biogenesis, through the enhancement of the nuclear translocation of TFEB and the downregulation of the mTORC1. -Ibudilast could prevent TDP-43-induced neurotoxicity. | [81] |
| Multiple Sclerosis | In vivo | EAE rat models. | -Ibudilast pretreatment could prevent EAE in rats, although it could not alter the clinical course in case it was administered after the onset of the disease. -Ibudilast pretreatment could reduce neuroinflammatory responses in the spinal cord, inhibit MBP-induced T cell proliferation in the lymph nodes, reduce release of IFN-γ from T cells, and decrease secretion of TNF-α from macrophages. | [93] |
| Wolfram syndrome | In vitro | Rat insulinoma (INS1) cells. | -Knock out of WFS1 resulted in increased resting cytosolic calcium levels, downregulation of calcium signaling, and reduced insulin secretion. -Ibudilast and calpain inhibitor XI could also restore calcium homeostasis, cell viability and insulin secretion. | [101] |
| Glaucoma | In vivo | Rat models of ocular hypertension. | -Intraocular administration of ibudilast was associated with reduced microglia activation in the retina and optic nerve, resulting in reduced pro-inflammatory cytokines and gliosis, increased survival and restored axonal degeneration, via the upregulation of cAMP/PKA signaling pathway. | [105] |
| Neurodegenerative Disease | Clinical Trial | Study Design | Study Objectives | Main Findings | Reference |
|---|---|---|---|---|---|
| Amyotrophic Lateral Sclerosis | NCT02238626 | Randomized placebo-controlled Phase 1b/2a clinical trial | To evaluate the tolerability, safety, and clinical efficacy of ibudilast (60 mg/day) as an adjunct therapy to the standard riluzole treatment | -In the early cohort, ibudilast was safe and well-tolerated over a twelve-month period. -No significant difference in clinical progression was detected between ibudilast and placebo groups, as assessed by ALSFRS-R, hand-held dynamometry and ALSAQ-5. -Subgroup analysis demonstrated that ibudilast might provide more benefit for ALS patients with upper limb or bulbar onset, and possibly delay the progression of the disease if administered at an early stage, particularly if the onset of symptoms at screening was less than 17.1 months. | [35,82] |
| NCT02714036 | Open-label phase 1b clinical trial | To measure the impact of ibudilast on inflammation and axonal loss | -Ibudilast (up to 100 mg/day) was ineffective in inhibiting microglia activation in the primary motor cortex of ALS patients as evaluated by PBR28-PET over 12–24 weeks, and serum neurofilament light chain (NfL) levels, an indicator of neuronal axonal loss, remained unchanged over 36–40 weeks. -Most participants experienced at least one possibly ibudilast-related adverse event: about one-third of the patients required dosage reduction, while about another one-third discontinued ibudilast treatment because of ibudilast-related adverse events. | [49] | |
| NCT04057898 | Phase 2b/3 randomized, double-blind, placebo-controlled clinical trial | To evaluate the safety, tolerability, and efficacy of ibudilast (up to 100 mg/day) for twelve months, followed by an open-label extension phase for six months in patients with ALS | Ongoing | ||
| Progressive Multiple Sclerosis | NCT01982942 | Phase 2 randomized placebo-controlled clinical trial | To evaluate the safety, tolerability, and activity of ibudilast administered twice daily over a 96- week period in subjects with primary or secondary progressive multiple sclerosis | -Ibudilast (up to 100 mg/day) over a period of 96 weeks was associated with slower progression of the whole-brain atrophy and gray matter atrophy of patients with primary and secondary progressive MS. -Ibudilast was not associated with fewer new or enlarging T2-weighted or new T1-weighted MRI lesions. -Ibudilast treatment could also potentially attenuate retinal thinning on OCT. -Disability progression was similar between the ibudilast and placebo groups. -No significant alterations in NfL levels in the serum and CSF between ibudilast and placebo groups. -Most common adverse events in this study were gastrointestinal complains, headache, and depressive symptoms. -The overall treatment effect of ibudilast in brain atrophy was mainly driven by patients with primary progressive MS and not secondary progressive MS. | [18,36,95,96,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelopoulou, E.; Pyrgelis, E.-S.; Piperi, C. Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence. Molecules 2022, 27, 8448. https://doi.org/10.3390/molecules27238448
Angelopoulou E, Pyrgelis E-S, Piperi C. Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence. Molecules. 2022; 27(23):8448. https://doi.org/10.3390/molecules27238448
Chicago/Turabian StyleAngelopoulou, Efthalia, Efstratios-Stylianos Pyrgelis, and Christina Piperi. 2022. "Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence" Molecules 27, no. 23: 8448. https://doi.org/10.3390/molecules27238448
APA StyleAngelopoulou, E., Pyrgelis, E.-S., & Piperi, C. (2022). Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence. Molecules, 27(23), 8448. https://doi.org/10.3390/molecules27238448