
Development of Phosphoramidite Reagents for the Synthesis of Base-Labile Oligonucleotides Modified with a Linear Aminoalkyl and Amino-PEG Linker at the 3′-End


Abstract
:
1. Introduction
2. Results and Discussion
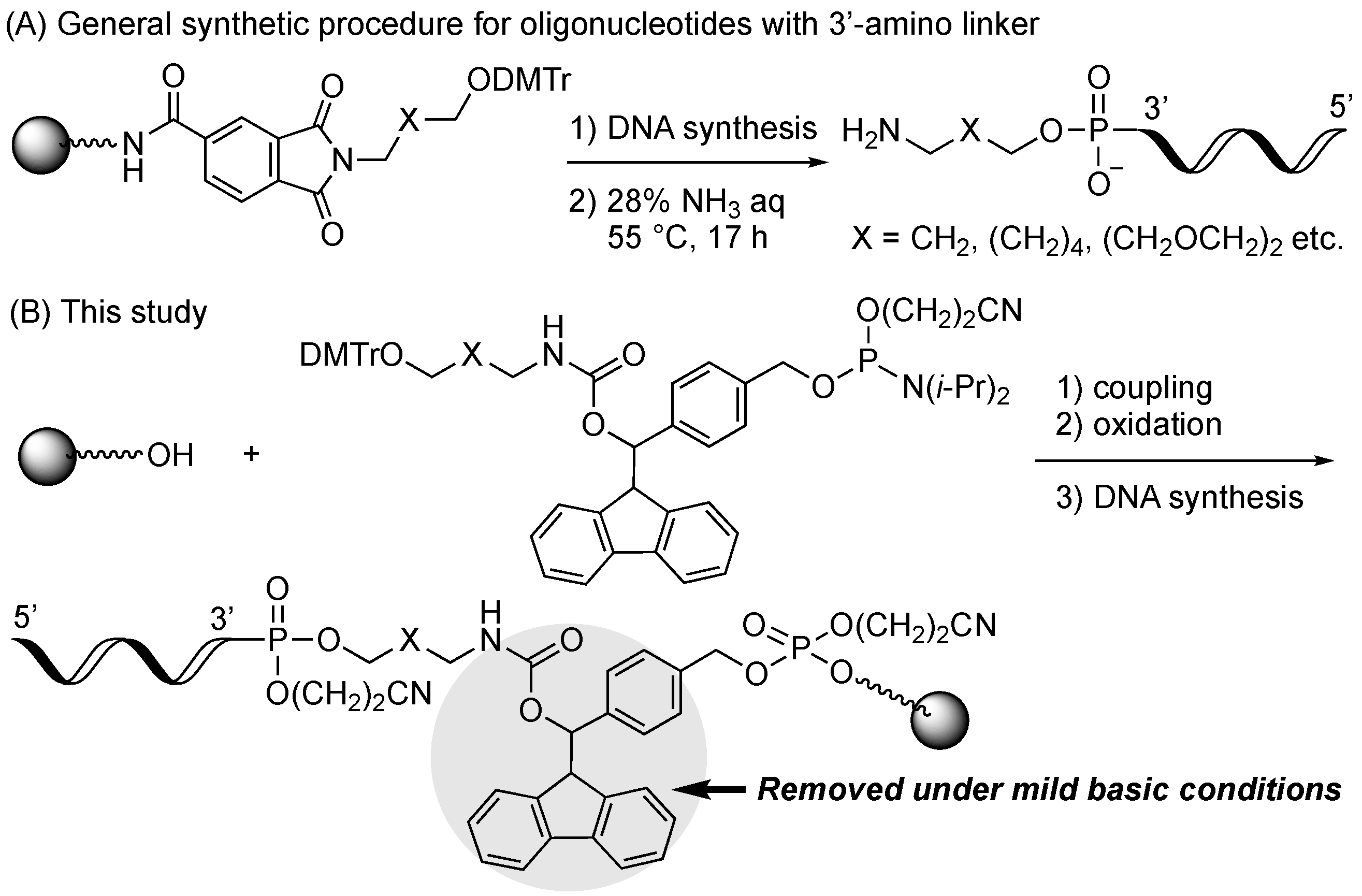
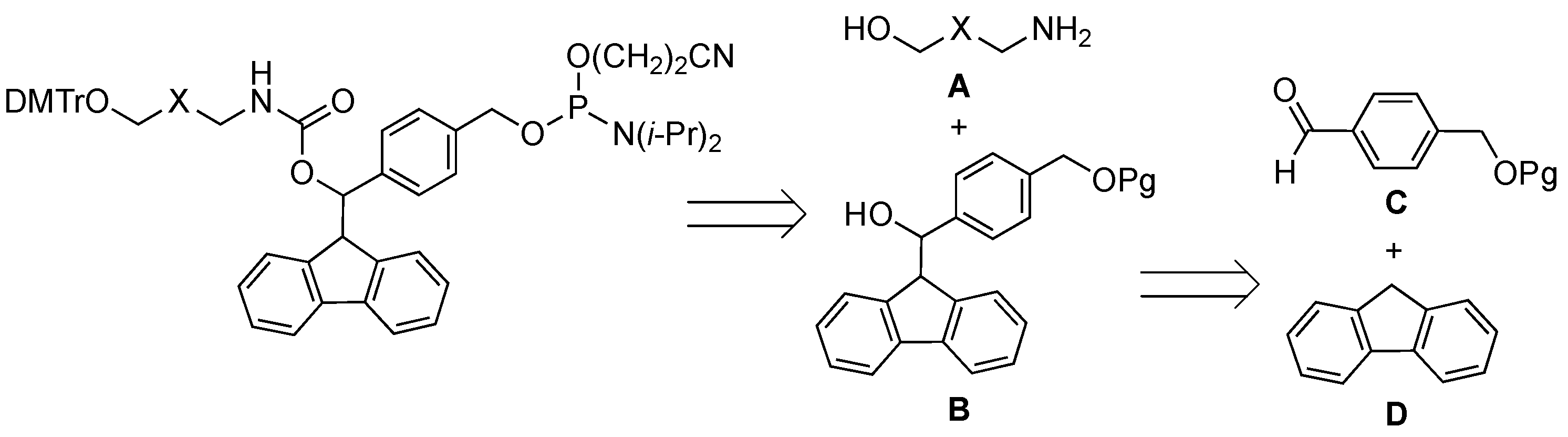
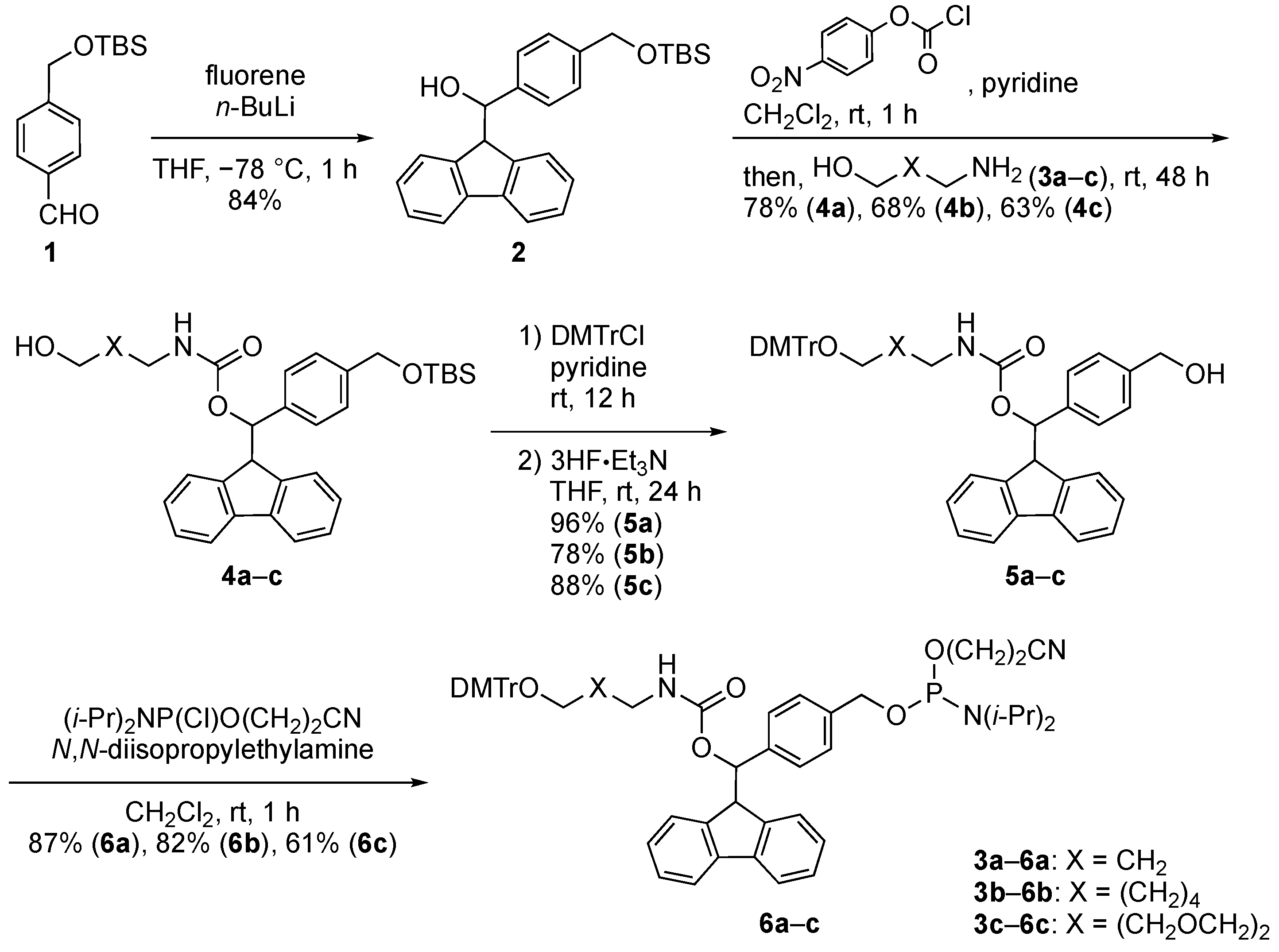
2.1. Synthiesis of Phosphoramidite Reagents
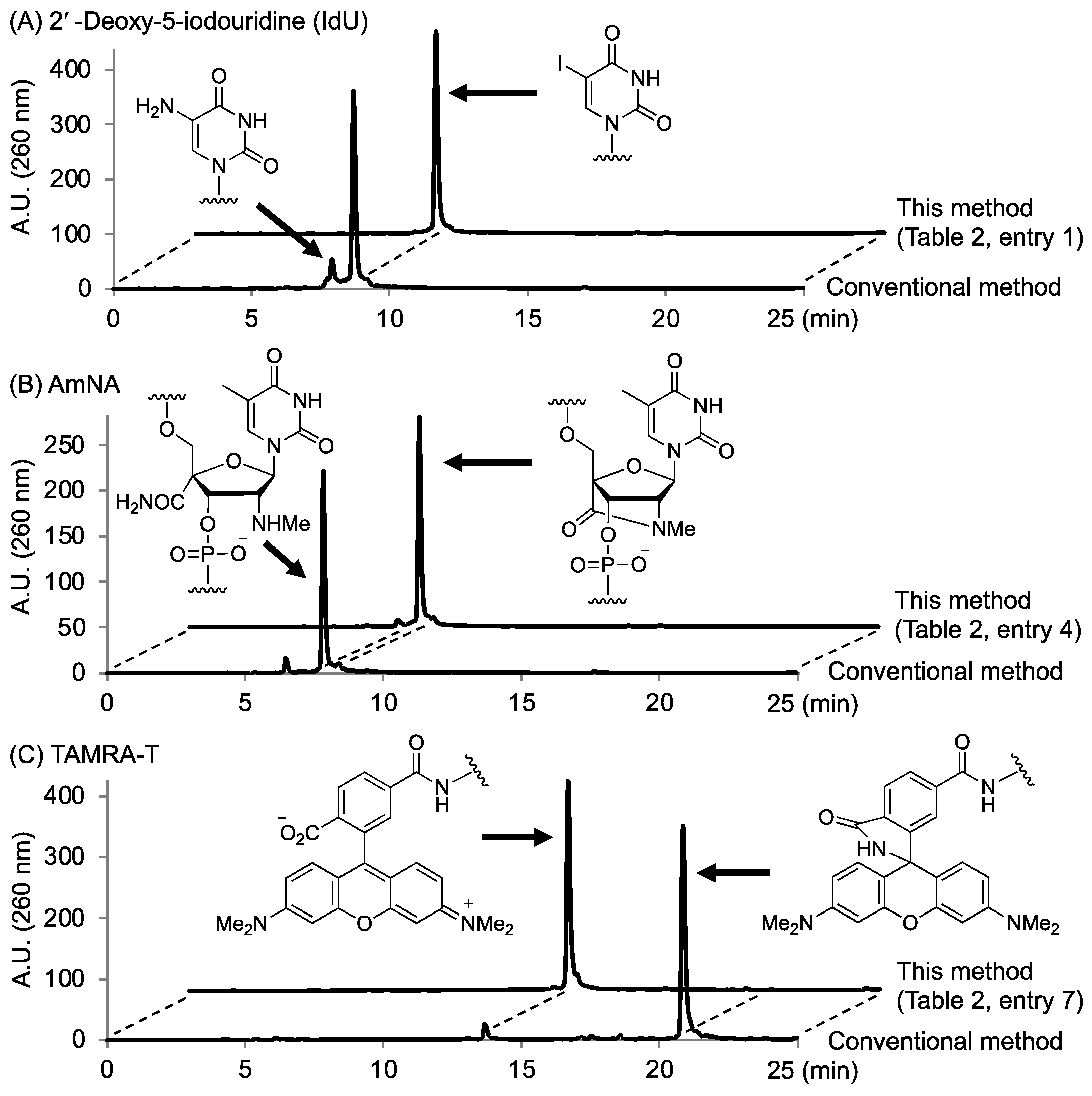
2.2. Optimum Deprotection Conditions for Oligonucleotides Modified with the Developed Phosphoramidite Reagents
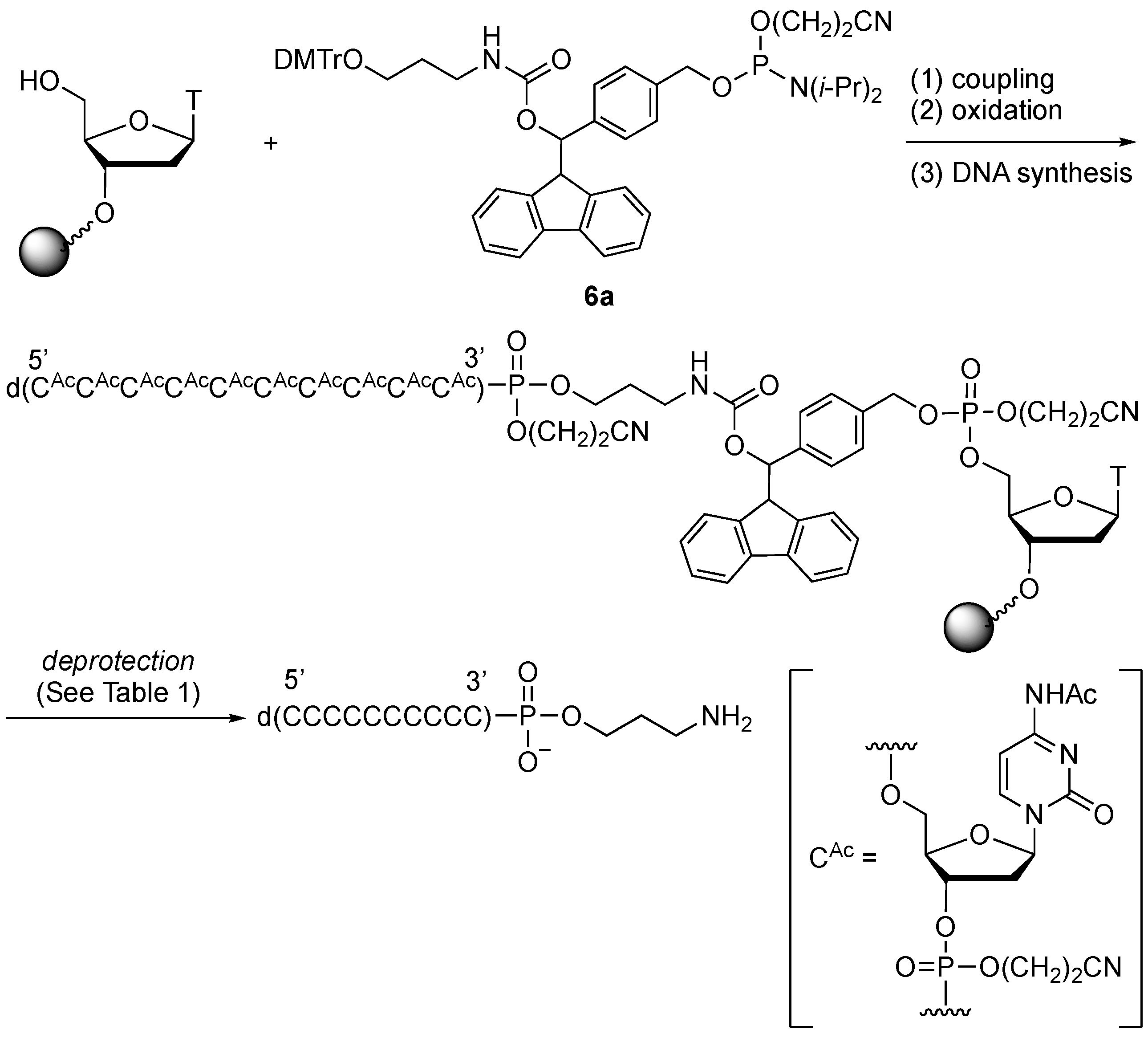
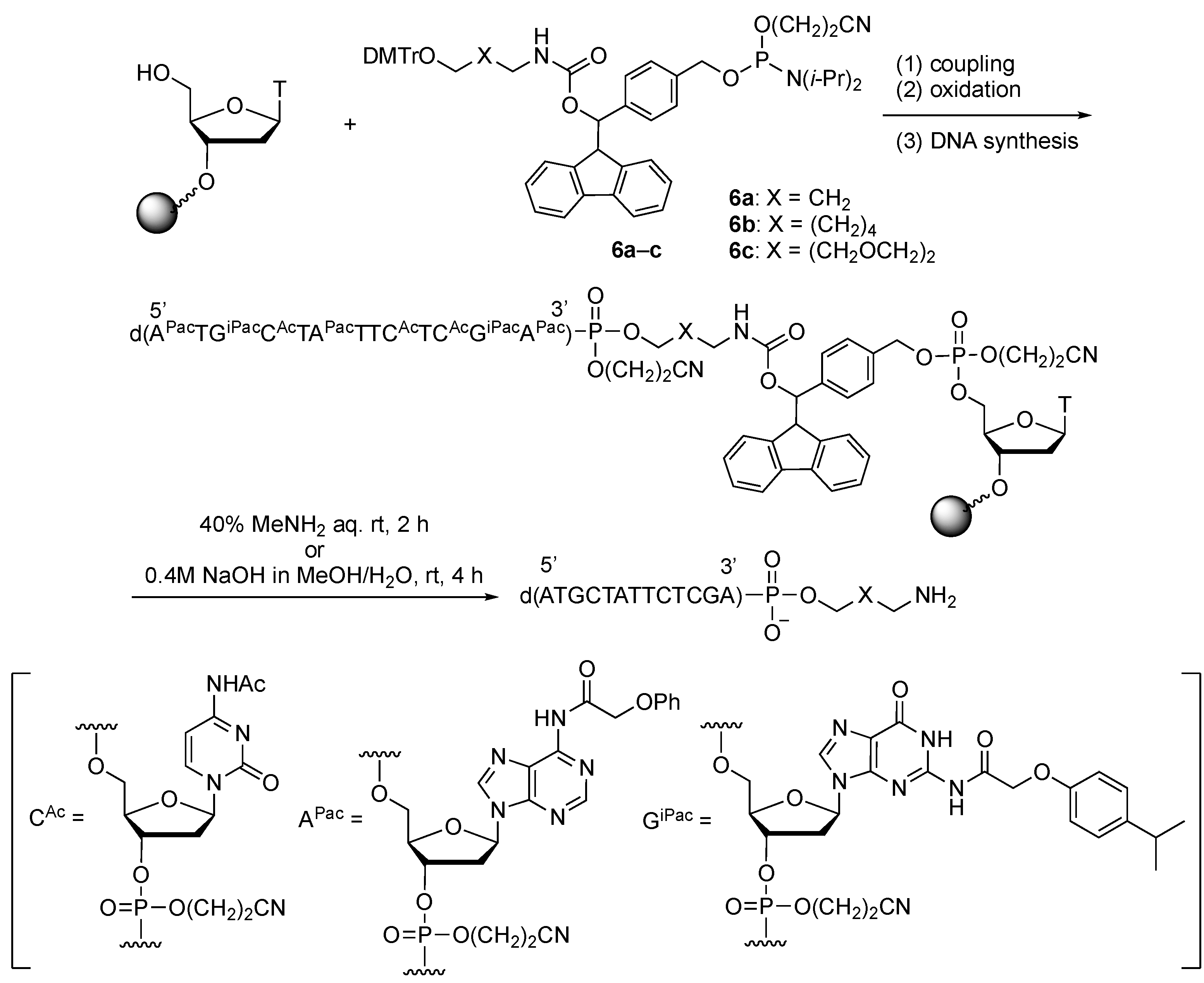
2.3. Synthesis of Oligonucleotides Modified with 3′-Amino Linkers and Base-Labile Nucleosides
3. Materials and Methods
3.1. General
3.2. Synthesis of Phosphoramidite Reagents 6a–c
3.3. Synthesis of Oligonucleotides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, H.; Zhu, X.; Li, S.; Wang, P.; Fang, J. Liver-targeted delivery of oligonucleotides with N-acetylgalactosamine conjugation. ACS Omega 2021, 6, 16259–16265. [Google Scholar] [CrossRef] [PubMed]
- Oyama, S.; Yamamoto, T.; Yamayoshi, A. Recent advances in the delivery carriers and chemical conjugation strategies for nucleic acid drugs. Cancers 2021, 13, 3881. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug derively. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Fissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Bartrhelemy, P. Bioconjugated oligonucleotides: Recent developments and therapeutic applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wu, Y.; Yu, J.; Zhang, W.; Zhuang, C. DNA-encoded libraries (DELs): A review of on-DNA chemistries and their output. RSC Adv. 2021, 11, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Gironda-Martínez, A.; Donckele, E.J.; Samain, F.; Neri, D. DNA-encoded chemical libraries: A comprehensive review with successful stories and future challenges. ACS Pharmacol. Transl. Sci. 2021, 4, 1265–1279. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, P.; Su, J.; Zhang, N.; Gu, H.; Zhao, Y. DNA nanotechnology-facilitated ligand manipulation for targeted therapeutics and diagnostics. J. Contr. Rel. 2021, 340, 292–307. [Google Scholar] [CrossRef]
- Huang, Z.; Qiu, L.; Zhang, T.; Tan, W. Integrating DNA nanotechnology with aptamers for biological and biomedical applications. Matter 2021, 4, 461–489. [Google Scholar] [CrossRef]
- Whitfield, C.J.; Zhang, M.; Winterwerber, P.; Ng, D.Y.W.; Weil, T. Functional DNA-polymer conjugates. Chem. Rev. 2021, 121, 11030–11084. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Yu, J.; Kinberger, G.A.; Wan, W.B.; Migawa, M.T.; Vasquez, G.; Schmidt, K.; Gaus, H.J.; Murray, H.M.; Low, A.; et al. Efficient synthesis and biological evaluation of 5′-GalNAc conjugated antisense oligonucleotides. Bioconjug. Chem. 2015, 26, 1451–1455. [Google Scholar] [CrossRef]
- Li, Y.; Gabriele, E.; Samain, F.; Favalli, N.; Sladojevich, F.; Scheuermann, J.; Neri, D. Optimized reaction conditions for amide bond formation in DNA-encoded combinatorial libraries. ACS Comb. Sci. 2016, 18, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.; Pedroso, E.; Grandas, A. Conjugation reactions involving maleimides and phosphorothioate oligonucleotides. Bioconjug. Chem. 2012, 23, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.; Pedroso, E.; Grandas, A. Easy introduction of maleimides at different positions of oligonucleotide chains for conjugation purposes. Org. Biomol. Chem. 2012, 10, 8478–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, A.; Vasseur, J.J.; Morvan, F. Synthesis of monoconjugated and multiply conjugated oligonucleotides by “click thiol” thiol-Michael-type additions and by combination with CuAAC “click Huisgen”. Eur. J. Org. Chem. 2013, 2013, 465–473. [Google Scholar] [CrossRef]
- Bouillon, C.; Meyer, A.; Vidal, S.; Jochum, A.; Chevolot, Y.; Cloarec, J.P.; Praly, J.P.; Vasseur, J.J.; Morvan, F. Microwave assisted ′′click′′ chemistry for the synthesis of multiple labeled- carbohydrate oligonucleotides on solid support. J. Org. Chem. 2006, 71, 4700–4702. [Google Scholar] [CrossRef]
- Gramlich, P.M.E.; Wirges, C.T.; Manetto, A.; Carell, T. Postsynthetic DNA modification through the copper-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem. Int. Ed. 2008, 47, 8350–8358. [Google Scholar] [CrossRef]
- Pourceau, G.; Meyer, A.; Vasseur, J.J.; Morvan, F. Azide solid support for 3′-conjugation of oligonucleotides and their circularization by click chemistry. J. Org. Chem. 2009, 74, 6837–6842. [Google Scholar] [CrossRef]
- Pourceau, G.; Meyer, A.; Vasseur, J.J.; Morvan, F. Synthesis of mannose and galactose oligonucleotide conjugates by bi-click chemistry. J. Org. Chem. 2009, 74, 1218–1222. [Google Scholar] [CrossRef]
- Busskamp, H.; Batroff, E.; Niederwieser, A.; Abdel-Rahman, O.S.; Winter, R.F.; Wittmann, V.; Marx, A. Efficient labelling of enzymatically synthesized vinyl-modified DNA by an inverse-electron-demand Diels-Alder reaction. Chem. Commun. 2014, 50, 10827–10829. [Google Scholar] [CrossRef] [Green Version]
- Weisbrod, S.H.; Marx, A. A nucleoside triphosphate for site-specific labelling of DNA by the Staudinger ligation. Chem. Commun. 2007, 18, 1828–1830. [Google Scholar] [CrossRef]
- Noël, M.; Clément-Blanc, C.; Meyer, A.; Vasseur, J.; Morvan, F. Solid support for the synthesis of 3′-aminooxy deoxy- or ribo-oligonucleotides and their 3′-conjugation by oxime ligation. J. Org. Chem. 2019, 84, 14853–14860. [Google Scholar] [CrossRef] [PubMed]
- Gamper, H.B.; Reed, M.W.; Cox, T.; Virosco, J.S.; Adams, A.D.; Gall, A.A.; Scholler, J.K.; Meyer Jr, R.B. Facile preparation of nuclease resistant 3′ modified oligodeoxynucleotides. Nucleic Acids Res. 1993, 21, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, C.R.; Reed, M.W.; Adams, A.D.; Meyer, R.B., Jr. An improved CPG support for the synthesis of 3′-amine-tailed oligonucleotides. Bioconjug. Chem. 1992, 3, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Lyttle, M.H.; Adams, H.; Hudson, D.; Cook, R.M. Versatile linker chemistry for synthesis of 3′-modified DNA. Bioconjug. Chem. 1997, 8, 193–198. [Google Scholar] [CrossRef] [PubMed]
- McMinn, D.; Greenberg, M.M. Postsynthetic conjugation of protected oligonucleotides containing 3′-alkylamines. J. Am. Chem. Soc. 1998, 120, 3289–3294. [Google Scholar] [CrossRef]
- Pourshahian, S. Therapeutic oligonucleotides, impurities, degradations, and their characterization by mass spectrometry. Mass Spectrom. Rev. 2021, 40, 75–109. [Google Scholar] [CrossRef]
- Capaldi, D.C. Stress testing of oligonucleotides. In Pharmaceutical Stress Testing: Predicting Drug Degradation, 2nd ed.; Baertschi, S.W., Alsante, K.M., Reed, R.A., Eds.; Informa Healthcare: Boca Raton, FL, USA, 2011; pp. 391–425. [Google Scholar]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [Green Version]
- Carpino, L.A.; Han, G.Y. The 9-fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J. Am. Chem. Soc. 1970, 92, 5748–5749. [Google Scholar] [CrossRef]
- Carpino, L.A.; Han, G.Y. The 9-fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972, 37, 3404–3409. [Google Scholar] [CrossRef]
- Smith, L.M.; Fung, S.; Hunkapiller, M.W.; Hood, L.E. The synthesis of oligonucleotides containing an aliphatic amino group at the 5′ terminus: Synthesis of fluorescent DNA primers for use in DNA sequence analysis. Nucleic Acids Res. 1985, 13, 2399–2412. [Google Scholar] [CrossRef]
- Nelson, P.S.; Kent, M.; Muthini, S. Oligonucleotide labeling methods 3. Direct labeling of oligonucleotides employing a novel, non-nucleosidic, 2-aminobutyl-1,3-propanediol backbone. Nucleic Acids Res. 1992, 20, 6253–6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chládek, S.; Romano, L.J. Synthesis of oligonucleotides containing site-specific carcinogen adducts. Preparation of the 2-cyanoethyl N,N-diisopropylphosphoramidite of N-(2′-deoxyguanosin-8-yl)-2-(acetylamino)fluorene with Fmoc as the base-protecting group. J. Org. Chem. 1994, 59, 556–563. [Google Scholar] [CrossRef]
- Johannsen, M.W.; Crispino, L.; Wamberg, M.C.; Kalra, N.; Wengel, J. Amino acids attached to 2′-amino-LNA: Synthesis and excellent duplex stability. Org. Biomol. Chem. 2011, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.; Vester, B.; Wengel, J. Oligonucleotides containing a piperazino-modified 2′-amino-LNA monomer exhibit very high duplex stability and remarkable nuclease resistance. Chem. Commun. 2015, 51, 4024–4027. [Google Scholar] [CrossRef] [Green Version]
- Lou, C.; Samuelsen, S.V.; Christensen, N.J.; Vester, B.; Wengel, J. Oligonucleotides containing aminated 2′-amino-LNA nucleotides: Synthesis and strong binding to complementary DNA and RNA. Bioconjug. Chem. 2017, 28, 1214–1220. [Google Scholar] [CrossRef]
- Danielsen, M.B.; Christensen, N.J.; Jørgensen, P.T.; Jensen, K.J.; Wengel, J.; Lou, C. Polyamine-functionalized 2′-amino-LNA in oligonucleotides: Facile synthesis of new monomers and high-affinity binding towards ssDNA and dsDNA. Chem. Eur. J. 2021, 27, 1416–1422. [Google Scholar] [CrossRef]
- Rydzik, A.M.; Balk, R.; Koegler, M.; Steinle, T.; Riether, D.; Gottschling, D. Access to-1′-amino carbocyclic phosphoramidite to enable postsynthetic functionalization of oligonucleotides. Org. Lett. 2021, 23, 6735–6739. [Google Scholar] [CrossRef]
- Panduwawala, T.D.; Iqbal, S.; Thompson, A.L.; Genov, M.; Pretsch, A.; Pretsch, D.; Liu, S.; Ebright, R.H.; Howells, A.; Maxwell, A.; et al. Functionalized bicyclic tetramates derived from cysteine as antibacterial agents. Org. Biomol. Chem. 2019, 17, 5615–5632. [Google Scholar] [CrossRef]
- Reddy, M.P.; Hanna, N.B.; Farooqui, F. Fast cleavage and deprotection of oligonucleotides. Tetrahedron Lett. 1994, 35, 4311–4314. [Google Scholar] [CrossRef]
- Miller, P.S.; Reddy, M.P.; Murakami, A.; Blake, K.R.; Lin, S.; Agris, C.H. Solid-phase synthesis of oligodeoxyribonucleotide methylphosphonates. Biochemistry 1986, 25, 5092–5097. [Google Scholar] [CrossRef]
- Reddy, M.P.; Farooqui, F.; Hanna, N.B. Methylamine deprotection provides increased yield of oligoribonucleotides. Tetrahedron Lett. 1995, 36, 8929–8932. [Google Scholar] [CrossRef]
- Lebedev, A.V.; Combs, D.; Hogrefe, R.I. Preactivated carboxyl linker for the rapid conjugation of alkylamines to oligonucleotides on solid support. Bioconjug. Chem. 2007, 18, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, T.; Yamaguchi, T.; Obika, S. Thioamide-bridged nucleic acid(thioAmNA) containing thymine or 2-thiothymine: Duplex-forming ability, base discrimination, and enzymatic stability. ChemBioChem 2019, 20, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Mullah, B.; Andrus, A. Automated synthesis of double dye-labeled oligonucleotides using tetramethylrhodamine (TAMRA) solid supports. Tetrahedron Lett. 1997, 38, 5751–5754. [Google Scholar] [CrossRef]
- Ferrer, E.; Wiersma, M.; Kazimierczak, B.; Müller, C.W.; Eritja, R. Preparation and properties of oligonucleotides containing 5-iodouracil and 5-bromo- and 5-iodocytosine. Bioconjug. Chem. 1997, 8, 757–761. [Google Scholar] [CrossRef]
- Yahara, A.; Shrestha, A.R.; Yamamoto, T.; Hari, Y.; Osawa, T.; Yamaguchi, M.; Nishida, M.; Kodama, T.; Obika, S. Amido-bridged nucleic acids (AmNAs): Synthesis, duplex stability, nuclease resistance, and in vitro antisense potency. ChemBioChem 2012, 12, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yahara, A.; Waki, R.; Yasuhara, H.; Wada, F.; Harada-Shiba, M.; Obika, S. Amido-bridged nucleic acids with small hydrophobic residues enhance hepatic tropism of antisense oligonucleotides in vivo. Org. Biomol. Chem. 2015, 13, 3757–3765. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagents | Temp. | Time | Deprotection Efficiency 1 |
|---|---|---|---|---|
| 1 | 28% NH3 aq. | 55 °C | 17 h | 100% |
| 2 | 28% NH3 aq. | rt | 2 h | 78% |
| 3 | 28% NH3 aq. | rt | 8 h | 100% |
| 4 | 40% MeNH2 aq. | rt | 2 h | 100% |
| 5 | 50 mM K2CO3 in MeOH | rt | 4 h | 55% |
| 6 | 50 mM K2CO3 in MeOH | rt | 24 h | 100% |
| 7 | 0.4 M NaOH in MeOH/H2O (4:1) | rt | 4 h | 100% |
| 8 | t-BuNH2/MeOH/H2O (1:1:2) | 65 °C | 3 h | 100% |

| Entry | X | Y | Reagents | Temp. | Time | Yield 1 |
|---|---|---|---|---|---|---|
| 1 | CH2 | IdU | 28% NH3 aq. | rt | 8 h | 33% |
| 2 | (CH2)4 | IdU | 28% NH3 aq. | rt | 8 h | 23% |
| 3 | (CH2OCH2)2 | IdU | 28% NH3 aq. | rt | 8 h | 31% |
| 4 | CH2 | AmNA | 50 mM K2CO3 in MeOH | rt | 24 h | 16% |
| 5 | (CH2)4 | AmNA | 50 mM K2CO3 in MeOH | rt | 24 h | 12% |
| 6 | (CH2OCH2)2 | AmNA | 50 mM K2CO3 in MeOH | rt | 24 h | 20% |
| 7 | CH2 | TAMRA-T | t-BuNH2/MeOH/H2O (1:1:2) | 65 °C | 3 h | 17% |
| 8 | (CH2)4 | TAMRA-T | t-BuNH2/MeOH/H2O (1:1:2) | 65 °C | 3 h | 12% |
| 9 | (CH2OCH2)2 | TAMRA-T | t-BuNH2/MeOH/H2O (1:1:2) | 65 °C | 3 h | 10% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osawa, T.; Ren, Q.; Obika, S. Development of Phosphoramidite Reagents for the Synthesis of Base-Labile Oligonucleotides Modified with a Linear Aminoalkyl and Amino-PEG Linker at the 3′-End. Molecules 2022, 27, 8501. https://doi.org/10.3390/molecules27238501
Osawa T, Ren Q, Obika S. Development of Phosphoramidite Reagents for the Synthesis of Base-Labile Oligonucleotides Modified with a Linear Aminoalkyl and Amino-PEG Linker at the 3′-End. Molecules. 2022; 27(23):8501. https://doi.org/10.3390/molecules27238501
Chicago/Turabian StyleOsawa, Takashi, Qin Ren, and Satoshi Obika. 2022. "Development of Phosphoramidite Reagents for the Synthesis of Base-Labile Oligonucleotides Modified with a Linear Aminoalkyl and Amino-PEG Linker at the 3′-End" Molecules 27, no. 23: 8501. https://doi.org/10.3390/molecules27238501
APA StyleOsawa, T., Ren, Q., & Obika, S. (2022). Development of Phosphoramidite Reagents for the Synthesis of Base-Labile Oligonucleotides Modified with a Linear Aminoalkyl and Amino-PEG Linker at the 3′-End. Molecules, 27(23), 8501. https://doi.org/10.3390/molecules27238501