Abstract
A new series of 1,2,3-triazole derivatives 5a–f based on benzothiazole were synthesized by the 1,3-dipolar cycloaddition reaction of S-propargyl mercaptobenzothiazole and α-halo ester/amide in moderate to good yields (47–75%). The structure of all products was characterized by 1H NMR, 13C NMR, and CHN elemental data. This protocol is easy and green and proceeds under mild and green reaction conditions with available starting materials. The structural and electronic analysis and 1H and 13C chemical shifts of the characterized structure of 5e were also calculated by applying the B3LYP/6-31 + G(d, p) level of density functional theory (DFT) method. In the final section, all the synthesized compounds were evaluated for their anti-inflammatory activity by biochemical COX-2 inhibition, antifungal inhibition with CYP51, anti-tuberculosis target protein ENR, DPRE1, pks13, and Thymidylate kinase by molecular docking studies. The ADMET analysis of the molecules 5a–f revealed that 5d and 5a are the most-promising drug-like molecules out of the six synthesized molecules.
1. Introduction
Among the types of heterocycles, systems containing nitrogen, oxygen, and sulfur atoms have the most activity and biological aspects [1]. So far, many of these organic compounds that have pharmacological activity have been synthesized and used [2]. Five-membered rings with two carbon and three nitrogen atoms are called triazoles. Triazoles have two types of isomers: 1,2,3-triazole and 1,2,4-triazole. Using a 1,3-dipolar cycloaddition reaction between alkynes and aryl/alkyl azides, 1,2,3-triazoles can be easily synthesized as two isomers of 1,4- and 1,5-disubstituted triazoles [3,4,5,6]. In 2002, Meldal and Sharples showed that the 1,3-dipolar cycloaddition reaction between an azide and terminal alkyne in the presence of copper (I) resulted in the regioselective synthesis of 1,4 disubstituted-1,2,3-triazoles [7,8]. 1,2,3-Triazoles have been used to prepare commercial and clinical drugs [9,10]. Some of the biological properties of these compounds are anti-epileptic, anti-cancer, anti-viral, anti-microbial, anti-fungal, anti-tumor, anti-diabetic, anti-tuberculosis, anti-inflammatory, anti-HIV, and anti-allergic [10,11,12,13,14,15,16]. The application of benzotriazole derivatives as polymer field effect transistor materials was also reported [17]. Benzothiazoles are also vital heterocyclic compounds found in many natural products. Several clinical benzothiazole-based drugs have been found to be effective in treating various diseases. These compounds have medicinal and biological properties, including anti-cancer, anti-tuberculosis, anti-HIV, anti-fungal, anti-bacterial, anti-diabetic, anti-malarial, anti-inflammatory, antioxidant, and antihistamine [18,19,20,21,22]. Benzothiazoles have many applications in polymers and dyes [23,24,25]. 2-Mercaptobenzothiazole acts as an accelerator in rubber vulcanization [26].
Due to the importance and application of 1,2,3-triazoles bearing the benzothiazole group [27,28], the synthesis of these derivatives is essential for organic chemists and biologists, and efforts are underway to design drugs based on these compounds with high activity and lower toxicity. As a result of our group’s experience synthesizing 1,4-disubstituted 1,2,3-triazole derivatives [29,30,31,32,33,34], the new 1,2,3-triazoles were prepared using benzothiazole groups 5a–f via a 1,3-dipolar cycloaddition of S-propargylated mercaptobenzothiazole 3, sodium azide, and α-halo ester/amide 4 in the presence of CuSO4/sodium ascorbate in a mixture of H2O/t-BuOH (Scheme 1). The density functional theory (DFT) calculations, with the B3LYP/6-31 + G(d, p) method, were applied to investigate the physicochemical properties of the characterized structure of 5e, including structural, electronic, and spectral analysis. Moreover, the study concludes by examining each of the synthesized compounds to specific targets using molecular docking to predict the possible biological activity of the proposed molecules. As mentioned above, triazoles have anti-inflammatory, anti-tuberculosis, and anti-fungal properties. For this purpose, the COX-2 protein was chosen for the anti-inflammatory target. COX-2 has been an important target for pain reduction drugs through the years and even shows some evidence relating this protein to cancer [35,36,37]. Numerous studies have been conducted on this protein with triazole structures in silico, in vitro, and in vivo that have shown promising results [38,39]. For anti-tuberculosis activity, four different targets were selected. DPRE1 has been one of the potential targets for resistant tuberculosis. Anti-tuberculosis target protein ENR was another protein target on our list. The thymidylate kinase of Mycobacterium tuberculosis and Pks13 are the other anti-tuberculosis targets we selected for this research due to their critical role in this disease. For anti-fungal activity, we analyzed our synthesized molecule’s activity against Saccharomyces cerevisiae CYP51 [40,41,42,43,44,45,46,47,48,49,50,51]. Absorption, distribution, metabolism, excretion, and toxicities (ADMET) prediction is one of the essential steps for drug discovery and development. After the docking study, we analyzed the molecules 5a–f for ADMET properties to analyze and predict these molecules against the control molecules for each target to better understand the chances of introducing them to the next phase of drug discovery and development. Our molecular docking experiments for the molecule 5e, which has a chiral center, were conducted to check how the enantiomers affect its potential activity and interactions with proteins. Due to the limitations of ADMET servers, which predict properties based on the 2D structure of molecules, we could not generate separate properties for each of the two enantiomers.
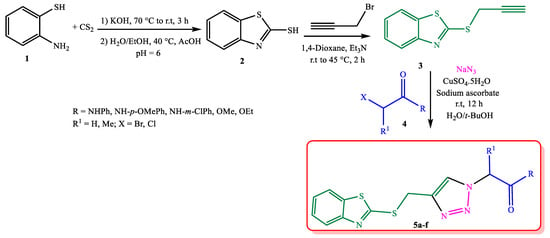
Scheme 1.
Efficient synthesis of novel 1,2,3-triazoles bearing the benzothiazole group 5a–f.
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterization of 1,2,3-Triazoles 5a–f
2-Mercaptobenzothiazole 2 was obtained according to the literature [52] and then was propargylated by propargyl bromide at 45 °C in dioxane in the presence of Et3N (Scheme 1). The azide intermediates were obtained by the in situ reaction of the corresponding α-halo ester/amide 4 with sodium azide. The desired 1,2,3-triazoles 5a–f were prepared by the azide-alkyne [3 + 2] cycloaddition reaction at room temperature. To optimize the reaction conditions, the solvent, catalyst, and temperature effect were investigated on the model reaction of 3 with 4a in the presence of sodium azide (Table 1). It should be mentioned that increasing the temperature had no good effect on the yield of the reaction (Entries 1–3 and 5, Table 1). With the inclusion of a low amount of copper catalyst (30 or 10 mol%), the reaction took longer, as well as resulted in a low yield of the product. CuSO4 (50 mol%) as a catalyst, sodium ascorbate as a reductant agent (50 mol%), and H2O/t-BuOH (1:1) as solvent at room temperature were applied as the best reaction conditions (Entry 4, Table 1).

Table 1.
Optimization of catalyst, solvent, and temperature for the synthesis of 5a a.
The generality of the protocol was then studied. As shown in Scheme 2, 1,2,3-triazole derivatives (5a–f) were synthesized in moderate to good yields (47–75%) by the reaction of a series of α-halo ester/amide 4 and S-propargyl mercaptobenzothiazole 3. The structure of triazoles 5a–f was established based on 1H NMR, 13C NMR spectral data, and CHN analysis. As a representative example, the 1H NMR and 13C NMR data of the product 5b are discussed. The 1H NMR spectrum of 5b consisted of a singlet peak at δ = 3.72 ppm for the hydrogens of the methoxy group, a broad line at δ = 4.75 ppm for the methylene of the -CH2S- group, a broad line at δ = 5.27 ppm for –CH2CO-, eight aromatic hydrogens at δ = 6.91–8.04 ppm, a sharp characteristic signal at δ = 8.16 ppm for hydrogen on the triazole ring, and a broad line at δ = 10.34 ppm correlating with –CONH (amidic hydrogen). The amidic hydrogen (acceptor) (N-H…N) forms a strong intramolecular hydrogen bond with the nitrogen atom of the triazole ring (donor), resulting in a deshielded proton for the amide moiety in 1H NMR. The 1H-decoupled 13 CNMR spectrum of 5b showed 17 distinct resonances, a resonance at δ = 166.30 ppm for the carbon of carbonyl group, 13 distinct resonances for aromatic carbons between δ = 114.44–164.02 ppm, two peaks at δ = 55.60 and 52.61 ppm for the carbons of the methylene groups, and a resonance at δ = 27.85 ppm for the carbon of the methoxy group, which is in agreement with the proposed structure.
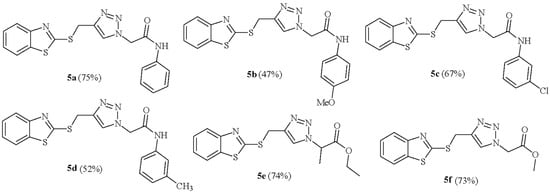
Scheme 2.
The structure of the synthesized 1,2,3-triazole derivatives 5a–f.
2.2. Theoretical Studies on the 1,2,3-Triazole 5e
In continuation, the physicochemical properties of 5e, such as structural, electronic, and spectral data, were investigated by the DFT-B3LYP computational method in conjunction with the 6–31 + G(d, p) basis set.
2.2.1. Benchmarking of the Computational Method
DFT calculations have successfully provided the theoretical background of popular qualitative chemical concepts [53]. Furthermore, organic chemists use the DFT-B3LYP method for various chemical calculations [54,55,56,57]. This method has been utilized in the present study for performing calculations. The experimental 1H NMR and 13C NMR data for 5e could pave the way to detect the accurate computational method.
The experimental 1H NMR spectrum of 5e consisted of a triplet at δ = 1.10 for methyl, a doublet for methyl at 1.72, a multiplet at 4.41 for methylene (-OCH2-), and a singlet at 4.71 for methylene (-SCH2−), a quartet at 5.62 for –NCH- (1H), four protons for phenyl rings at δ = 7.35–7.99 ppm, and a characteristic signal at δ = 8.24 ppm for hydrogen on the triazole ring. The 1H-decoupled 13C NMR spectrum of 5e showed 15 resonances, which is in agreement with the proposed structure, while the carbonyl appearing at δ = 169.21 ppm, nine distinct resonances for the aromatic carbons of phenyl and triazole rings between δ = 121.30–165.74 ppm, and five resonances at δ = 13.80–61.66 ppm for aliphatic carbons. The simulated NMR spectra for 5e were obtained at the B3LYP/6-31 + G(d, p) level with the aid of the Gauss View software [58]. Table 2 summarizes the calculated and experimental 1H and 13C chemical shifts for 5e. The 1H and 13C chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane as the reference, and its corresponding shifts were calculated at the same theoretical level. For the 6-31 + G(d, p)-optimized geometry of 5e in DMSO, the NMR chemical shifts were also calculated using the same basis set and solvent as the experimental NMR spectra.
Table 2.
B3LYP/6-31 + G(d, p) calculated and experimental 1H and 13C chemical shifts (in ppm) of 5e in DMSO.
Table 2.
B3LYP/6-31 + G(d, p) calculated and experimental 1H and 13C chemical shifts (in ppm) of 5e in DMSO.
| Atom a | δB3LYP | δexp | Atom a | δB3LYP | δexp | Atom a | δB3LYP | δexp |
|---|---|---|---|---|---|---|---|---|
| H35−37 | 1.27 | 1.01 | H19 | 8.15 | 8.01 | C4 | 119.53 | 121.82 |
| H27−29 | 1.83 | 1.72 | H7 | 8.17 | 8.24 | C6 | 120.86 | 123.85 |
| H32 | 4.10 | 4.10 | C34 | 16.54 | 13.80 | C18 | 123.18 | 124.59 |
| H16 | 4.55 | 4.71 | C26 | 20.67 | 17.05 | C5 | 123.45 | 126.41 |
| H25 | 5.55 | 5.62 | C15 | 35.50 | 27.44 | C2 | 139.12 | 134.76 |
| H10 | 7.62 | 7.35 | C24 | 64.14 | 57.53 | C20 | 140.05 | 142.29 |
| H9 | 7.77 | 7.49 | C31 | 66.31 | 61.66 | |||
| H8 | 8.09 | 7.90 | C1 | 118.96 | 120.31 |
a For the numbering of atoms, refer to Figure 1.

Figure 1.
B3LYP/6-31 + G(d, p) optimized geometry of 5e in DMSO.
Figure 1.
B3LYP/6-31 + G(d, p) optimized geometry of 5e in DMSO.

The calculated 1H and 13C chemical shifts are in excellent agreement with the experimental data; regression was obtained R2 = 0.996 for 1H NMR and R2 = 0.995 for 13C NMR data (Figure 2). 1H NMR comparison showed a better correlation rather than 13C NMR. Based on the results, it is possible to continue the calculations and obtain the physicochemical properties of 5e using the B3LYP/6–31 + G(d, p) method.
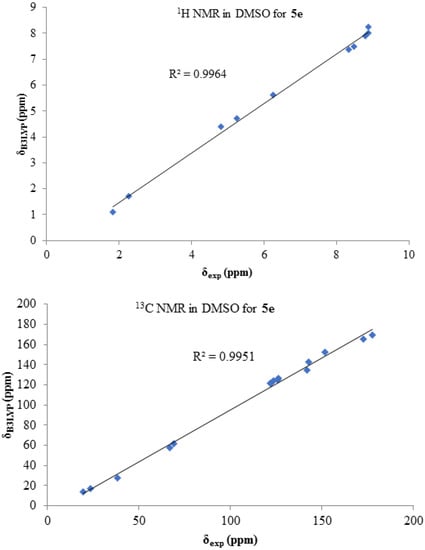
Figure 2.
1H and 13C chemical shifts’ correlation between calculated and experimental data for 5e in DMSO.
2.2.2. Structural Characterization of 5a and 5e
Figure 1 illustrates the optimized geometry of 5e at the B3LYP/6-31 + G(d, p) computational level in DMSO. Table 3 shows the representative calculated bond lengths and bond angles for 5e, which are within normal ranges. The bond C = O was 1.215 Å. The single S-C bonds extend in the range of 1.757–1.856 Å. The C20-C18-N23 and C1-C2-N13 angles are 104.6 and 115.3°. In the esteric group, C24-C30-O38 was calculated as 109.7°, while the O39-C30-O38 resulted as 125.5°.
Table 3.
The calculated structural parameters of 5e by the B3LYP/6-31 + G(d, p) method.
As mentioned above, there is strong intramolecular hydrogen bonding (IMHB) between the amidic hydrogen and nitrogen atom of the triazole ring (N-H…N) in the chemical structures of 5a–d. This interaction was confirmed from the 1H NMR spectra of 5a–d, resulting in a deshielded proton for the amide group. As can be seen from the optimized structures of 5a (Figure 3), there is a very strong IMHB with the hydrogen atom of the amide group as the acceptor center and the nitrogen atom of the triazole ring as the donor site. By analyzing the natural bond orbital (NBO) of 5a, we can gain valuable insight into the intermolecular/intramolecular interactions, such as hydrogen bonding, numerically according to two-order perturbation energy (E(2)) [59]. The LP (1) N22 → BD*(1) N28-H41 as a remarkable interaction with energy of 4.85 kcal mol−1 in 5a is attributed to the IMHB interaction. The structures of 5b–d exhibit also an analogous trend.
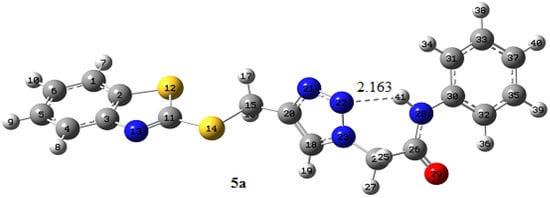
Figure 3.
The B3LYP/6-31 + G(d, p) optimized geometry of 5a (distance in Å).
2.2.3. Electronic Characterization of 5e
Table 4 shows the electronic data calculated by the B3LYP/6-31 + G(d, p) method of 5e in DMSO. The dipole moment in DMSO for 5e is 5.81 Debye, indicating the high polarity. The presence of polar groups, such as the amide group and unsymmetrical structure, are the reasons for this high dipolar moment data.
Table 4.
The electronic data at the B3LYP/6-31 + G(d, p) level for 5e in DMSO.
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the frontier orbitals that reveal the ability to donate or accept an electron, respectively. It is worth mentioning that removing electrons from HOMO is easier, and this orbital can donate electrons. As shown in Figure 4, the HOMO and LUMO orbitals are localized mainly on the benzothiazole ring. A useful parameter for determining the organic compound’s reactivity is the gap between the HOMO and LUMO energy levels (Eg). The difference between EHOMO and ELUMO (Eg) is 4.98 eV (Table 4).
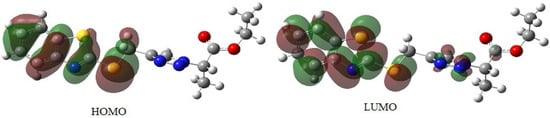
Figure 4.
HOMO and LUMO for 5e calculated by B3LYP/6-31 + G(d, p) level.
The global electrophilicity index (GEI) is a criterion of the molecule’s ability to accept electrons, which has been used to determine some adequate information on the structure, reactivity, dynamics, aromaticity, Lewis acidity, and toxicity [60,61]. Parr defined the GEI (ω) as [62]:
where μ and η are the chemical potential and hardness, respectively, given by Equation (2); 5e has a GEI of 2.94 eV. The GEI can be categorized based on Domingo’s scale [63]. Electrophiles are divided into three groups based on the GEI value: strong, mild, and weak. When the electrophilic index ω is more than 1.5 eV, the electrophile is strong; in the range of 0.8 to 1.5 eV, it is mild, and when it is less than 0.8 eV, it is a weak electrophile. Based on Domingo’s scale, the compound 5e is a strong electrophile and can easily interact with a typical nucleophile. The NBO charge distribution for the structure of 5e is shown in Figure 5. According to the NBO analysis, there is a remarkable excess of positive charge (1.466 e) on the carbon atom of the carbonyl group. The study also indicates that the carbon atom of the imine unit (N = C) in the benzothiazole ring, methylene (-SCH2-), and –NCH- bear a positive charge of 1.106, 0.338, and 0.421e, respectively. NBO analysis and the electrophilicity index of 5e show that a nucleophile can attack these positive carbon centers. Possible nucleophilic centers in 5e, based on NBO analysis and the electrophilicity index, are shown in Figure 5.
ω = μ2/2η
μ = (ELUMO + EHOMO)/2 and η = (ELUMO − EHOMO)/2
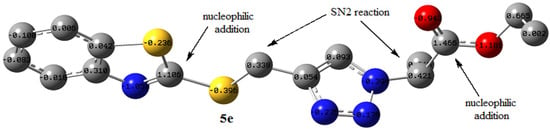
Figure 5.
Possible nucleophilic sites in 5e, based on NBO analysis and the electrophilicity index (hydrogen atoms are omitted for clarity).
2.3. Molecular Docking and ADMET Studies
Molecular docking results show that molecule 5d has the highest binding energy for 1G3U (−9.7 kcal mol−1), 1QSG (−9.1 kcal mol−1), and 5F19 (−9.6 kcal mol−1), even more than the control molecules. Based on this information, 5d is likely to be a promising inhibitor of the proposed target. Molecule 5b has the same binding energy as 5d for 1QSG, giving the idea of the potential anti-tuberculosis activity of this molecule. In 4ZE2, the binding energy of the control molecule is similar to 5a, equal to −9.7 kcal mol−1. The synthesized molecules 5a–f show higher binding energy and more minor interactions with two other proteins (4P8N, 5V3X), indicating that these molecules have a lower anti-tuberculosis activity than their reference molecules. In 4P8N, the control molecule BTZ043 shows a binding energy of −10.6 kcal mol−1. In addition, 5d has the lowest binding energy of all proposed molecules, with a binding energy of −9.6 kcal mol−1. As shown by the binding energy of 5V3X, the reference molecule has a binding energy of −10.1 kcal mol−1, which indicates a strong interaction between this molecule and the protein target. Given that the six synthesized molecules do not bypass the reference molecule and have lower inhibitory activity against the target, 5d has the best binding energy of −9.7 kcal mol−1. A summary of the docking results can be found in Table 5.
Table 5.
Binding energy data for the triazoles 5a–f and control molecules (in kcal mol−1).
All compounds 5a–f can bind to the active site of the protein targets. The synthesized compounds can form hydrogen bonds (H-bond) with the amino acids in the active site of the protein targets. The number of H-bonds of 5a–f with the related protein targets is collected in Table 6. H-bond interactions were found between nitrogen atoms on the triazole ring of the compounds 5a–f and amino acids in the active site of the protein targets, indicating that the triazole unit of the synthesized compounds could be the key to binding and inhibiting the protein (Figure 6).
Table 6.
A summary of the ADMET properties for 5a–f and the control molecules.
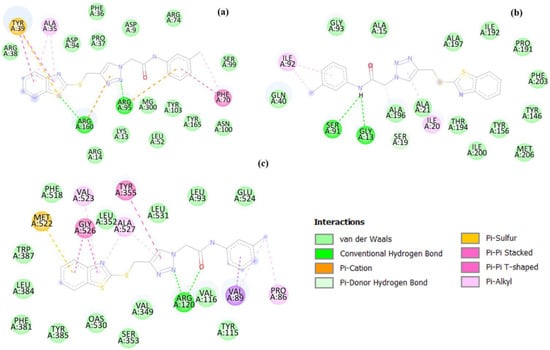
Figure 6.
Two-dimensional illustrations of the interaction types between 5d and protein targets, 1G3U (a), 1QSG (b), and 5F19 (c).
It is interesting to note that the compound 5e has two enantiomers (5e-R and 5e-S). Molecular docking studies of these enantiomers can be interesting. Except for the protein 1QSG, the binding energy of 5e-S with other proteins is higher than 5e-R (Table 6). For example, we considered only the interaction of 5e-R and 5e-S with 1G3U and 4P8N. For 1G3U, the 5e-R interacts with ARG-160 through a hydrogen bond, one carbon–hydrogen interaction with PRO-37, six van der Waals interactions with SER-99, TYR-103, ASP-9, MG-300, ARG-14, and ALA-35, and several other interactions such as P–cation, Pi–anion, Pi–sulfur, Pi–Pi stacked, and Pi–alkyl. 5e-S shows more interactions, specifically four hydrogen bonds with LYS-13, ARG-95, TYR-39, and ARG-160. Moreover, it also has a carbon–hydrogen bond with ASP-9. The 5e-S molecule also has eight van der Waals interactions with ARG-153, ARG-14, ASP-94, ARG-74, SER-99, ASN-100, LEU-52, and ASP-162. Like 5e-R, 5e-S also interacts with the active site, interactions such as Pi–cation, P–anion, Pi–sulfur, Pi–Pi stacked, Pi–Pi T-shaped, and Pi–alkyl. These results could indicate the higher binding affinity of 5e-S in comparison to 5e-R (Figure 7). Based on the interaction results for these enantiomers for 4P8N, 5e-R has two hydrogen bonds with LYS-134 and LYS-418 and six alkyl and Pi–alkyl interactions with PRO-316, CYS-387, TYR314, ILE-131, and PRO-116. There are also a Pi–donor hydrogen bond with GLY-117 and nine van der Waals interaction with TYR-415, HIS-132, GLY-133, GLN-336, THR-118, VAL-121, ALA-417, LYS-367, and SER-228. Moreover, the 5e-R molecule has a Pi–sigma interaction with VAL-365. Molecule 5e-S has one hydrogen bond with ARG-58 and eight alkyl and Pi–alkyl interactions with MET-74, ILE-184, ALA-128, ILE-131, PRO-116, CYX-129, and TYR-415. Furthermore, there are one carbon–hydrogen bond with GLY-125 and thirteen van der Waals interaction with LYS-418, ILE-183, ALA-94, THR-122, GLY-55, ASN-63, ARG-54, ALA-126, VAL-121, ALA-417, GLY-57, ALA-64, and GLY-179. These are some other interactions that 5e-S creates with the protein (Figure 7).
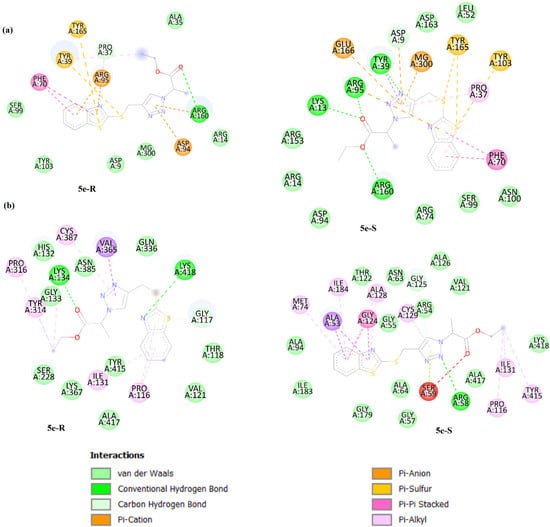
Figure 7.
Two-dimensional illustrations of the interaction types between 5e-R and 5e-S and protein targets, 1G3U (a) and 4P8N (b).
The Lipinski, Pfizer, and GSK rules are the focus of the ADMET study. While the molecules 5a, 5b, and 5d showed the best docking results, the ADMET analysis reveals that all three mentioned molecules passed two out of the three rules above. The control molecule for 4ZE2, Posaconazole, passes only the Pfizer rule, suggesting that the molecules 5a, 5b, and 5d have a better chance than others, mainly molecule 5d, owing to its robust docking against anti-inflammatory and anti-tuberculosis targets. The ADMET results are summarized in Table 6.
Lipophilicity (permeability of a drug to reach the target tissue in the body) and solubility are two important molecular properties for the absorption of a drug. The calculated solubility in water (Log S) of the synthesized compounds 5a–f followed the order: 5f > 5e > 5a > 5b > 5d > 5c. The estimated lipophilicity (Log P) also followed the order: 5c > 5d > 5b > 5a > 5e > 5f. After analyzing the chemical structures of 5a–f (Scheme 2), it is easy to understand that, as the number of hydrocarbon chains increases, the solubility in water decreases and lipophilicity increases.
3. Experimental Section
3.1. General Information
The chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO. USA), Fluka (Neu-Ulm, Germany), and Merck (Darmstadt, Germany). The structure of the synthesized triazole derivatives 5a–f was confirmed by 1H NMR, 13C NMR, and CHN elemental data. A Bruker (DRX-500 Avance) NMR was used to record the 1H and 13C NMR spectra in CDCl3 and DMSO at room temperature (The Supplementary Material). The multiplicity of the proton signals is abbreviated with s for singlet, d for doublet, m for multiplet, and dd for the doublet of doublet peaks. Quantitative microanalyses (CHN) of the products 5a–f were carried out on a Thermo Finnigan Flash-1112EA microanalyzer.
3.2. Computational Details
Geometric optimization of the characterized structure 5e was performed at the DFT/B3LYP level using the 6-31 + G(d, p) basis set utilizing the Gaussian 09 software [64]. The vibrational frequencies were calculated simultaneously to ensure that each stationary point was a real minimum. NMR data calculations were performed using the gauge-independent atomic orbital (GIAO) method [65]. Relative chemical shifts were calculated using the corresponding tetramethylsilane for 1H NMR and 13C NMR shielding, calculated at the same level of theory as the reference. Further, the solvent effects were taken into account by using the conductor-like polarizable continuum model (CPCM) [66] through DFT calculations performed at the B3LYP/6-31 + G(d, p) level of theory in DMSO as the solvent.
3.3. Molecular Docking and ADMET Details
Molecular docking is a crucial component of the in silico study of drug discovery and development. We performed in silico docking studies with Pyrx to demonstrate the synthesized compounds’ anti-tuberculosis, anti-fungal, and anti-inflammatory properties. Pyrx was used to determine the interactions between the studied compounds and the specific protein targets [67]. All the molecules’ smiles were extracted by Cheminfo [68]. Afterward, all the structures were minimized using chimera and saved as PDB files. The next step involved unifying the six PDB structures using OpenBabel and converting them to a single SDF file [69]. RSCB PDB was used to extract all protein structures. Chimera was applied in all protein structures to remove solvents, water molecules, unnecessary ions, and ligands [70]. We selected a single control molecule per protein target for molecular docking to compare our synthesized molecules 5a–f with the reference molecules. All the procedures to prepare the reference molecules for docking were the same as for 5a–f, as shown above. Each target protein’s PDB ID and the control molecules are listed below in Table 7. Pyrx was then used to dock the preprocessed data. For the ADMET approach, we used the ADMETlab2 web server to analyze and predict the synthesized molecules’ properties [71]. In this section, all synthesized molecules 5a–f and control molecules are converted into their smile strings and fed to ADMETlab2.
Table 7.
Protein names, the related PDB ID, and their control molecules for docking study.
3.4. General Synthesis Procedure for 2-Mercaptobenzothiazole 2
To a solution of 2-aminothiophenol 1 (1 mmol) in 30 mL of KOH solution (10%), CS2 (6 mmol) was added and refluxed for three hours. The mixture was cooled, then 25 mL water and 25 mL ethanol were added and heated until 40 °C. At this temperature, 4.5 mL acetic acid was added dropwise to the reaction mixture to adjust the pH = 6. The mixture was cooled to room temperature, filtrated, and dried overnight to obtain the desired 2-mercaptobenzothiazole 2 in 90% yield.
3.5. General Synthesis Procedure for S-Propargylated Mercaptobenzothiazole 3
To a solution of 2-mercaptobenzothiazole 2 (5 mmol) in 10 mL dioxane, Et3N (5 mmol) was added and stirred at room temperature for 45 min. Propargyl bromide (5 mmol, 80% in toluene) was added dropwise to the reaction mixture and refluxed at 45 °C for one hour. After completion of the reaction, the mixture was cooled, and then, 20 mL of water was added. The reaction mixture was extracted by DCM (3 × 5 mL). The organic layer was dried over Na2SO4, filtrated, and evaporated to give the desired S-propargyl mercaptobenzothiazole 3 in 95% yield.
3.6. General Synthesis Procedure for 1,2,3-Triazole Derivatives 5a–f
To a solution of t-BuOH/H2O (1:1, 5 mL), α-halo ester/amide 4 (1 mmol) and NaN3 (1.3 mmol) were added and stirred at room temperature for one hour, then CuSO4 and sodium ascorbate (50% mol) and S-propargyl mercaptobenzothiazole 3 (1 mmol) were added. The reaction mixture was stirred at room temperature for 12 h. After completion of the reaction, as monitored by TLC, 10 mL of water was added, and the mixture was extracted with DCM (3 × 5 mL). The organic layer was dried over Na2SO4, filtrated, and evaporated under a vacuum. The desired products 5a–f were purified by column chromatography using ethyl acetate/hexane (1:1). The structure of the synthesized triazole derivatives 5a–f was confirmed by 1H and 13C NMR spectra data and CHN analysis.
4. Conclusions
We successfully reported synthesizing a series of new 1,2,3-triazoles based on benzothiazole by the 1,3-dipolar cycloaddition reaction of S-propargyl mercaptobenzothiazole and α-halo ester/amide in moderate to good yields. This protocol is easy and green and proceeds under mild reaction conditions with available starting materials. We also studied the full structural, electronic, and NMR data of the synthesized compound 5e using the B3LYP/6-31 + G(d, p) computational method. Molecular docking and ADMET studies indicate that the 5d scaffold could be the key to binding with the protein targets more than the other five synthesized molecules in terms of their anti-inflammatory and anti-tuberculosis activity. All synthesized molecules did not achieve a better docking result in terms of antifungal activity than the reference molecules; however, 5a has the same binding affinity for 4ZE2 as the control molecule, suggesting that this molecule may possess antifungal properties. Further in vitro analysis is needed to prove the in silico results.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27238555/s1, Spectral data of the 1,2,3-triazoles 5a–f.
Author Contributions
Z.M.: Conceptualization, formal analysis, investigation, resources, software, validation, visualization, writing—review and editing. M.M.K.: formal analysis, investigation, resources, validation, visualization. P.H.: conceptualization, formal analysis, investigation, resources, software, validation, visualization, writing—review and editing. H.R.S.: investigation, resources, validation, visualization. A.A.: advising, writing—review and editing. H.S.: conceptualization, formal analysis, investigation, resources, software, validation, visualization, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Jampilek, J. Heterocycles in medicinal chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Arora, V.; Lamba, H.S.; Wadhwa, D. Importance of heterocyclic chemistry: A review. Int. J. Pharm. Sci. 2012, 3, 2947–2954. [Google Scholar]
- Breugst, M.; Reissig, H.U. The Huisgen Reaction: Milestones of the 1, 3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef]
- Matin, M.M.; Matin, P.; Rahman, M.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 303–310. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper (I)-catalyzed alkyne-azide cycloaddition (CuAAC)“click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Bedi, A.; Manor Armon, A.; Diskin-Posner, Y.; Bogosalvsky, B.; Gidron, O. Controlling the helicity of π-conjugated oligomers by tuning the aromatic backbone twist. Nat. Commun. 2022, 13, 451. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1, 2, 3]-triazoles by regiospecific copper (I)-catalyzed 1, 3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1, 2, 3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Dixit, D.; Verma, P.K.; Marwaha, R.K. A review on ‘triazoles’: Their chemistry, synthesis and pharmacological potentials. J. Iran. Chem. Soc. 2021, 18, 2535–2565. [Google Scholar] [CrossRef]
- Tratrat, C.; Haroun, M.; Paparisva, A.; Geronikaki, A.; Kamoutsis, C.; Ćirić, A.; Glamočlija, J.; Soković, M.; Fotakis, C.; Zoumpoulakis, P.; et al. Design, synthesis and biological evaluation of new substituted 5-benzylideno-2-adamantylthiazol [3, 2-b][1, 2, 4] triazol-6 (5H) ones. Pharmacophore models for antifungal activity. Arab. J. Chem. 2018, 11, 573–590. [Google Scholar] [CrossRef]
- Sharma, M.K.; Parashar, S.; Chahal, M.; Lal, K.; Pandya, N.U.; Om, H. Antimicrobial and in-silico evaluation of novel chalcone and amide-linked 1, 4-disubstituted 1, 2, 3 triazoles. J. Mol. Struct. 2022, 1257, 132632. [Google Scholar] [CrossRef]
- Chu, X.M.; Wang, C.; Wang, W.L.; Liang, L.L.; Liu, W.; Gong, K.K.; Sun, K.L. riazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019, 166, 206–223. [Google Scholar] [CrossRef]
- Carvalho da Silva, F.D.; Cardoso, M.F.D.C.; Ferreira, P.G.; Ferreira, V.F. Biological Properties of 1H-1, 2, 3-and 2H-1, 2, 3-Triazoles. In Chemistry of 1, 2, 3-Triazoles; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2014; pp. 117–165. [Google Scholar]
- Fallah, Z.; Tajbakhsh, M.; Alikhani, M.; Larijani, B.; Faramarzi, M.A.; Hamedifar, H.; Mohammadi-Khanaposhtani, M.; Mahdavi, M. A review on synthesis, mechanism of action, and structure-activity relationships of 1, 2, 3-triazole-based α-glucosidase inhibitors as promising anti-diabetic agents. J. Mol. Struct. 2022, 1255, 132469. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole ring—A biologically active scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Bedi, A.; Senanayak, S.P.; Das, S.; Narayan, K.S.; Zade, S.S. Cyclopenta [c] thiophene oligomers based solution processable D–A copolymers and their application as FET materials. Polym. Chem. 2012, 3, 1453–1460. [Google Scholar] [CrossRef]
- Rana, A.; Siddiqui, N.; Khan, S.A. Benzothiazoles: A new profile of biological activities. Indian J. Pharm. Sci. 2007, 69, 10–17. [Google Scholar]
- Noolvi, M.N.; Patel, H.M.; Kaur, M. Benzothiazoles: Search for anticancer agents. Eur. J. Med. Chem. 2012, 54, 447–462. [Google Scholar] [CrossRef]
- Ali, R.; Siddiqui, N. Biological aspects of emerging benzothiazoles: A short review. J. Chem. 2013, 2013, 345198. [Google Scholar] [CrossRef]
- Gill, R.K.; Rawal, R.K.; Bariwal, J. Recent advances in the chemistry and biology of benzothiazoles. Arch. Pharm. 2015, 348, 155–178. [Google Scholar] [CrossRef]
- Kamal, A.; Syed, M.A.H.; Mohammed, S.M. Therapeutic potential of benzothiazoles: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Kabatc, J.; Jurek, K. Synthesis, properties, and application of new benzothiazole-based sensitisers in polymer chemistry. Color. Technol. 2015, 131, 183–191. [Google Scholar] [CrossRef]
- Henary, M.; Paranjpe, S.; Owens, E.A. Synthesis and applications of benzothiazole containing cyanine dyes. Heterocycl. Comm. 2013, 19, 1–11. [Google Scholar] [CrossRef]
- Debnath, S.; Bedi, A.; Zade, S.S. Thienopentathiepine: A sulfur containing fused heterocycle for conjugated systems and their electrochemical polymerization. Polym. Chem. 2015, 6, 7658–7665. [Google Scholar] [CrossRef]
- Fel’dshteĭn, M.S.; Eĭtingon, I.I.; Pevzner, D.M.; Strel’nikova, N.P.; Dogadkin, B.A. Derivatives of 2-Mercaptobenzothiazole and Dimethyldithiocarbamic Acid as Vulcanization Accelerators. Rubber Chem. Technol. 1959, 32, 983–991. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule Vikas, D.; Naveen; Lal, K. Synthesis, Antifungal Studies, Molecular Docking, ADME and DNA Interaction Studies of 4-Hydroxyphenyl Benzothiazole Linked 1,2,3-Triazoles. J. Mol. Struct. 2021, 1245, 131013. [Google Scholar] [CrossRef]
- Fahim, A.M.; Tolan, H.E.; El-Sayed, W.A. Synthesis of novel 1, 2, 3-triazole based acridine and benzothiazole scaffold N-glycosides with anti-proliferative activity, docking studies, and comparative computational studies. J. Mol. Struct. 2022, 1251, 131941. [Google Scholar] [CrossRef]
- Mirjafary, Z.; Ahmadi, L.; Moradi, M.; Saeidian, H. A copper (II)–thioamide combination as a robust heterogeneous catalytic system for green synthesis of 1, 4-disubstituted-1, 2, 3-triazoles under click conditions. RSC Adv. 2015, 5, 78038–78046. [Google Scholar] [CrossRef]
- Saeidian, H.; Sadighian, H.; Abdoli, M.; Sahandi, M. Versatile and green synthesis, spectroscopic characterizations, crystal structure and DFT calculations of 1, 2, 3-triazole-based sulfonamides. J. Mol. Struct. 2017, 1131, 73–78. [Google Scholar] [CrossRef]
- Saeidian, H.; Sadighian, H.; Arabgari, M.; Mirjafary, Z.; Ayati, S.E.; Najafi, E.; Moghaddam, F.M. Organocopper-based magnetically recoverable and reusable nanocatalyst for efficient synthesis of novel 1, 2, 3-triazole-based sulfonamides in green medium. Res. Chem. Intermed. 2018, 44, 601–612. [Google Scholar] [CrossRef]
- Paghandeh, H.; Saeidian, H. Expedient and click synthesis, spectroscopic characterizations and DFT calculations of novel 1, 5-bis (N-substituted 1, 2, 3-triazole) benzodiazepinedione scaffolds. J. Mol. Struct. 2018, 1157, 560–566. [Google Scholar] [CrossRef]
- Karbasi, M.M.; Mirjafary, Z.; Saeidian, H.; Mokhtari, J. Efficient synthesis and DFT analysis of novel 1, 2, 3-triazole-based dithiocarbamates. J. Mol. Struct. 2021, 1227, 129535. [Google Scholar] [CrossRef]
- Esmaeeli, Z.; Khodabakhshi, M.R.; Mirjafary, Z.; Saeidian, H. Efficient synthesis of novel 1, 2, 3-triazole-based diazepam derivatives by click CuAAC reaction: Spectroscopic characterizations and DFT studies. J. Mol. Struct. 2021, 1246, 131206. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 Promotes Tumor Growth and Suppresses Tumor Immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef]
- Chow, L.W.C.; Loo, W.T.Y.; Toi, M. Current Directions for COX-2 Inhibition in Breast Cancer. Biomed. Pharmacother. 2005, 59, 281–284. [Google Scholar] [CrossRef]
- Li, S.M.; Tsai, S.E.; Chiang, C.Y.; Chung, C.Y.; Chuang, T.J.; Tseng, C.C.; Jiang, W.P.; Huang, G.J.; Lin, C.Y.; Yang, Y.-C.; et al. New Methyl -5-(Halomethyl)-1-Aryl-1H-1,2,4-Triazole-3-Carboxylates as Selective COX-2 Inhibitors and Anti-Inflammatory Agents: Design, Synthesis, Biological Evaluation, and Docking Study. Bioorg. Chem. 2020, 104, 104333–104346. [Google Scholar] [CrossRef]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Gomaa, H.A.M.; Youssif, B.G.M.; Radwan, M.O.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. New Quinoline/1,2,4-Triazole Hybrids as Dual Inhibitors of COX-2/5-LOX and Inflammatory Cytokines: Design, Synthesis, and Docking Study. J. Mol. Struct. 2021, 1244, 130948. [Google Scholar] [CrossRef]
- Venkateswarlu, K.; Kelly, D.E.; Kelly, S.L. Characterization of Saccharomyces Cerevisiae CYP51 and a CYP51 Fusion Protein with NADPH Cytochrome P-450 Oxidoreductase Expressed in Escherichia Coli. Antimicrob. Agents Chemother. 1997, 41, 776–780. [Google Scholar] [CrossRef][Green Version]
- Baptista, R.; Bhowmick, S.; Shen, J.; Mur, L.A.J. Molecular Docking Suggests the Targets of Anti-Mycobacterial Natural Products. Molecules 2021, 26, 475. [Google Scholar] [CrossRef]
- Enugopala, K.N.; Chandrashekharappa, S.; Deb, P.K.; Tratrat, C.; Pillay, M.; Chopra, D.; Al-Shar’i, N.A.; Hourani, W.; Dahabiyeh, L.A.; Borah, P.; et al. Anti-Tubercular Activity and Molecular Docking Studies of Indolizine Derivatives Targeting Mycobacterial InhA Enzyme. J. Enzyme Inhib. Med. Chem. 2021, 36, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, M.A.V.; Mansour, R.A.; Atatreh, N.; Bryce, R.A. Analysis of Enoyl-Acyl Carrier Protein Reductase Structure and Interactions Yields an Efficient Virtual Screening Approach and Suggests a Potential Allosteric Site. Chem. Biol. Drug. Des. 2015, 87, 131–142. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Alian, A.; Ortiz de Montellano, P.R. Inhibition of the Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase InhA by Arylamides. Bioorg. Med. Chem. 2007, 15, 6649–6658. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, G.; Berakdar, N. Docking Studies of Flavonoid Compounds as Inhibitors of β-Ketoacyl Acyl Carrier Protein Synthase I (Kas I) of Escherichia Coli. J. Mol. Graph. Model. 2015, 61, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.M.; Parker, J.E.; Warrilow, A.G.S.; Rolley, N.J.; Kelly, S.L.; Kelly, D.E. Complementation of Saccharomyces Cerevisiae ERG11/CYP51 (Sterol 14α-Demethylase) Doxycycline-Regulated Mutant and Screening of the Azole Sensitivity of Aspergillus Fumigatus Isoenzymes CYP51A and CYP51B. Antimicrob. Agents Chemother. 2010, 54, 4920–4923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The Fungal CYP51s: Their Functions, Structures, Related Drug Resistance, and Inhibitors. Front. Microbiol. 2019, 10, 691–699. [Google Scholar] [CrossRef]
- Price, C.L.; Warrilow, A.G.S.; Parker, J.E.; Mullins, J.G.L.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Novel Substrate Specificity and Temperature-Sensitive Activity of Mycosphaerella Graminicola CYP51 Supported by the Native NADPH Cytochrome P450 Reductase. Appl. Environ. Microbiol. 2015, 81, 3379–3386. [Google Scholar] [CrossRef]
- Yadav, D.K.; Ahmad, I.; Shukla, A.; Khan, F.; Negi, A.S.; Gupta, A. QSAR and Docking Studies on Chalcone Derivatives for Antitubercular Activity AgainstM. TuberculosisH37Rv. J. Chemom. 2014, 28, 499–507. [Google Scholar] [CrossRef]
- Mir, F.; Shafi, S.; Zaman, M.S.; Kalia, N.P.; Rajput, V.S.; Mulakayala, C.; Mulakayala, N.; Khan, I.A.; Alam, M.S. Sulfur Rich 2-Mercaptobenzothiazole and 1,2,3-Triazole Conjugates as Novel Antitubercular Agents. Eur. J. Med. Chem. 2014, 76, 274–283. [Google Scholar] [CrossRef]
- Shafi, S.; Mahboob Alam, M.; Mulakayala, N.; Mulakayala, C.; Vanaja, G.; Kalle, A.M.; Pallu, R.; Alam, M.S. Synthesis of Novel 2-Mercapto Benzothiazole and 1,2,3-Triazole Based Bis-Heterocycles: Their Anti-Inflammatory and Anti-Nociceptive Activities. Eur. J. Med. Chem. 2012, 49, 324–333. [Google Scholar] [CrossRef]
- Ballabeni, M.; Ballini, R.; Bigi, F.; Maggi, R.; Parrini, M.; Predieri, G.; Sartori, G. Synthesis of Symmetrical N, N’-Disubstituted Thioureas and Heterocyclic Thiones from Amines and CS (2) over a ZnO/Al2O3 Composite as Heterogeneous and Reusable Catalyst. J. Org. Chem. Res. 1999, 64, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B. Conceptual density functional theory and some recent developments. Acta. Phys. Sin. 2009, 25, 590–600. [Google Scholar]
- Saeidian, H.; Sahandi, M. Comprehensive DFT study on molecular structures of Lewisites in support of the Chemical Weapons Convention. J. Mol. Struct. 2015, 1100, 486–495. [Google Scholar] [CrossRef]
- Ghiasi, M.; Oskouie, A.A.; Saeidian, H. Dynamic stereochemistry of Topiramate (anticonvulsant drug) in solution: Theoretical approaches and experimental validation. Carbohydr. Res. 2012, 348, 47–54. [Google Scholar] [CrossRef]
- Shams, B.; Saeidian, H. Design of the novel neutral organic superacids by comprehensive DFT study on organic fluorosulfuric acids. Comput. Theor. Chem. 2018, 1135, 48–55. [Google Scholar] [CrossRef]
- Saeidian, H.; Mirjafary, Z. Engineering non-ionic carbon super-and hyperbases by a computational DFT approach: Substituted allenes have unprecedented cation affinities. New J. Chem. 2020, 44, 12967–12977. [Google Scholar] [CrossRef]
- Dennington, R.I.I.; Keith, T.; Millam, J. GaussView, Version 4.1.2; Semichem Inc.: Shawnee Mission, KS, USA, 2007.
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Jupp, A.R.; Johnstone, T.C.; Stephan, D.W. The global electrophilicity index as a metric for Lewis acidity. Dalton Trans. 2018, 47, 7029–7035. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Roy, D.R.; Chattaraj, P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aizman, A.; Contreras, R. The electrophilicity index in organic chemistry. Comput. Theor. Chem. 2007, 19, 139–201. [Google Scholar]
- Frisch, J.M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E., Jr.; Burant, J.C.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2014, 1263, 243–250. [Google Scholar]
- Home Page of Cheminfo Website. Available online: https://www.cheminfo.org/ (accessed on 5 July 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, 5–14. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).