
Synthesis, Biological Evaluation, and Molecular Dynamics of Carbothioamides Derivatives as Carbonic Anhydrase II and 15-Lipoxygenase Inhibitors
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussions
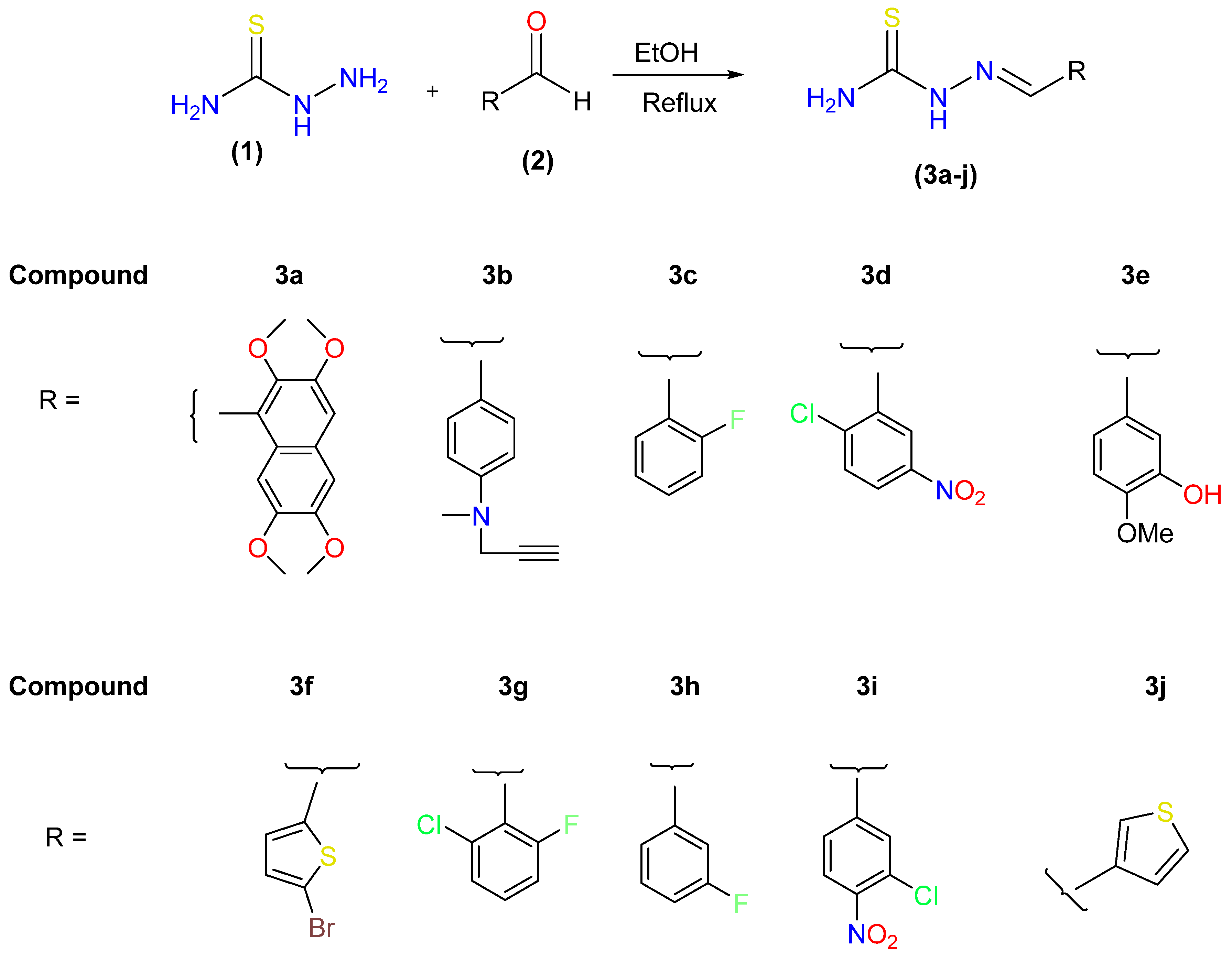
2.1. Chemistry
2.2. Carbonic Anhydrase and Lipoxygenase Activity and SAR
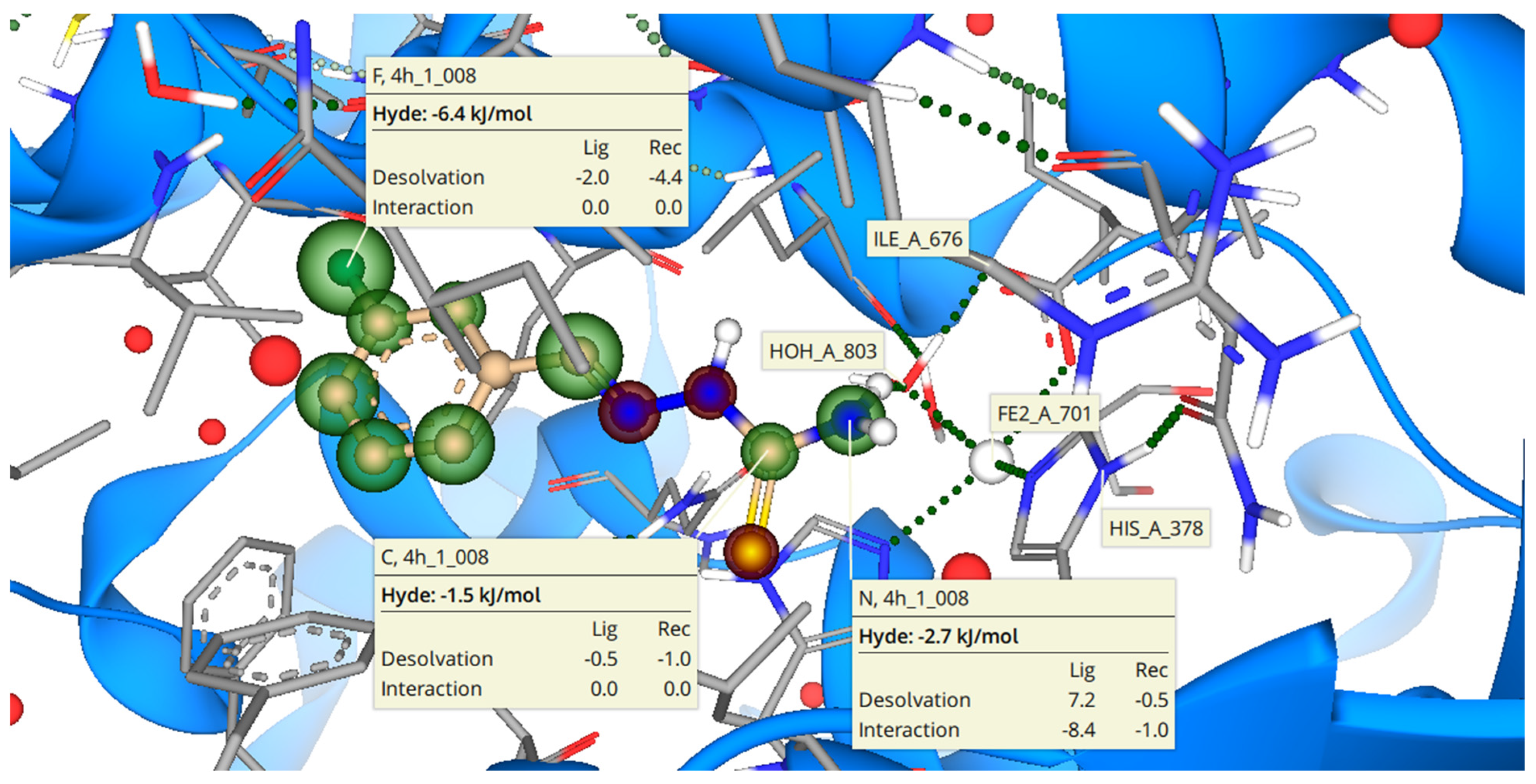
2.3. Binding Mode of 3h with b-CA II
2.4. Binding Mode of 3h with 15-LOX
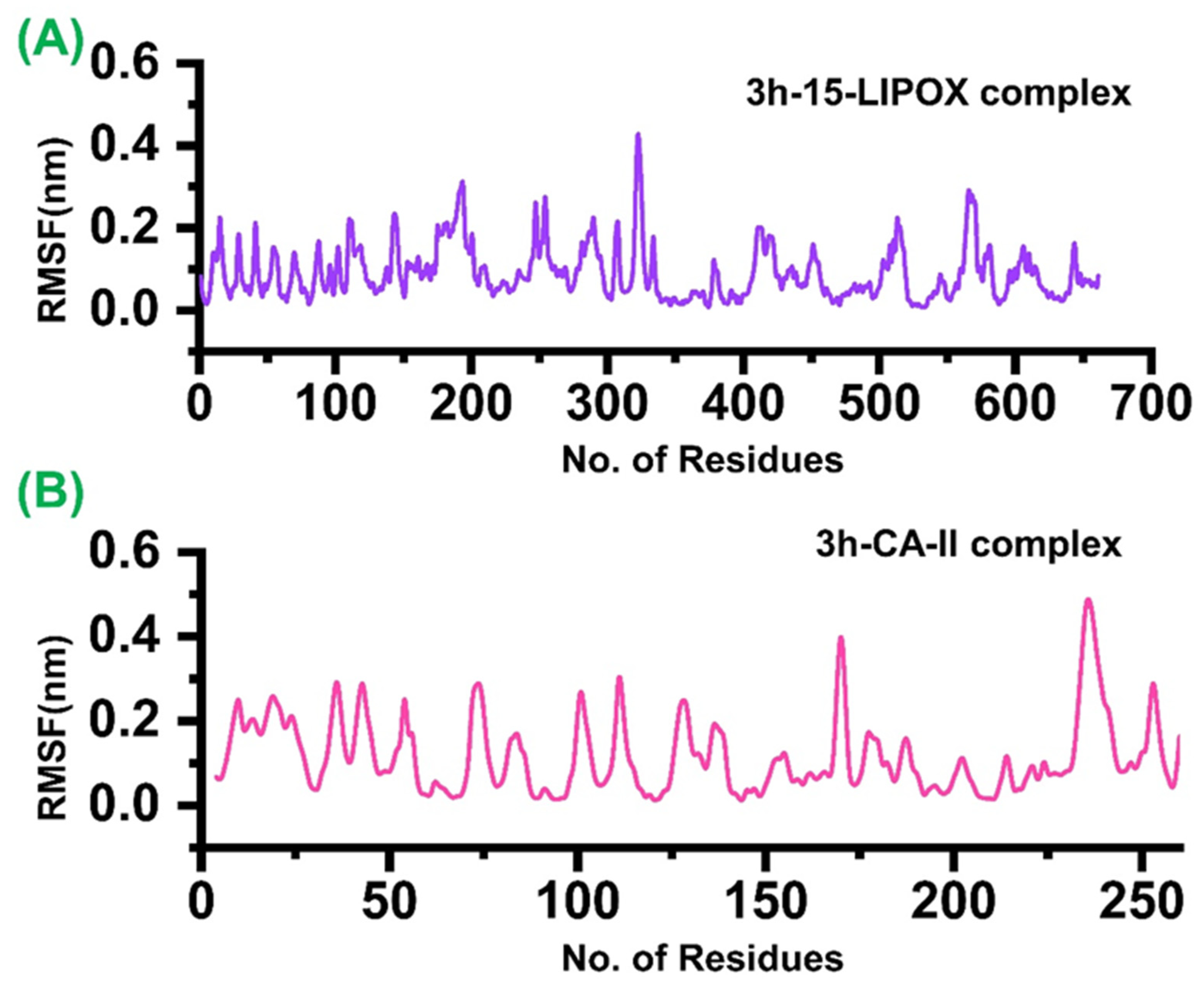
2.5. Dynamics Stability and Flexibility Profiling of the Two Ligand Bound Complexes
2.6. Binding Free Energy Calculation
3. Experimental
3.1. Characterization of Compounds
3.2. Synthesis of 2-(hetero (aryl) methylene) hydrazine-1carbothioamides (Schiff Bases) (3a–j)
3.3. Biochemical Assays
3.3.1. Lipoxygenase Assay
3.3.2. Carbonic Anhydrase Assay
3.4. Molecular Docking Studies
3.5. Molecular Dynamics Simulation
3.6. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matesanz, A.I.; Souza, P. α-N-heterocyclic thiosemicarbazone derivatives as potential antitumor agents: A structure-activity relationships approach. Mini-Rev. Med. Chem. 2009, 9, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Guo, C.; Hansell, E.; Doyle, P.S.; Caffrey, C.R.; Holler, T.P.; McKerrow, J.H.; Cohen, F.E. Synthesis and structure−activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 2002, 45, 2695–2707. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Z.-H.; Pan, S.-S.; Xu, H.-J.; Ren, J. Synthesis and antitumor activity of liquiritigenin thiosemicarbazone derivatives. Eur. J. Med. Chem. 2010, 45, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.C.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehrman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and structure−activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Thiele, B.J. The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett. 1999, 449, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Schewe, T. 15-lipoxygenase-1: A prooxidant enzyme. Biol. Chem. 2002, 383, 365–374. [Google Scholar] [CrossRef]
- Kelavkar, U.P.; Cohen, C. 15-Lipoxygenase-1 expression upregulates and activates insulin-like growth factor-1 receptor in prostate cancer cells. Neoplasia 2004, 6, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Camargo, A.B.; Marchevsky, E.; Luco, J.M. QSAR study for the soybean 15-lipoxygenase inhibitory activity of organosulfur compounds derived from the essential oil of garlic. J. Agric. Food Chem. 2007, 55, 3096–3103. [Google Scholar] [CrossRef]
- Dailey, L.; Imming, P. 12-Lipoxygenase: Classification, possible therapeutic benefits from inhibition, and inhibitors. Curr. Med. Chem. 1999, 6, 389–398. [Google Scholar] [CrossRef]
- Ding, X.-Z.; Tong, W.-G.; Adrian, T.E. Cyclooxygenases and lipoxygenases as potential targets for treatment of pancreatic cancer. Pancreatology 2001, 1, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, N.; Neochoritis, C.G.; Leus, N.G.; van der Wouden, P.E.; Domling, A.; Dekker, F.J. Rational development of a potent 15-lipoxygenase-1 inhibitor with in vitro and ex vivo anti-inflammatory properties. J. Med. Chem. 2015, 58, 7850–7862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S. Enhanced 15-lipoxygenase activity and elevated eicosanoid production in kidney tumor microenvironment contribute to the inflammation and immune suppression. Oncoimmunology 2012, 1, 249–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorekova, S.; Parkkila, S.; Parkkila, A.K.; Opavsky, R.; Zelnik, V.; Saarnio, J.; Pastorek, J. Carbonic anhydrase IX, MN/CA IX: Analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 1997, 112, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; He, Y.; Yan, G.; Hou, S.; Xie, Z.; Liao, C. Zinc enzymes in medicinal chemistry. Eur. J. Med. Chem. 2021, 226, 113877. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, N.; Wolff, M. Burger’s medicinal chemistry and drug discovery. In Therapeutic Agents, 5th ed.; Wolff, M.E.J., Ed.; Wiley Sons: New York, NY, USA, 1996. [Google Scholar]
- Supuran, C.; Scozzafava, A. Carbonic anhydrase and their therapeutic potentials. Exp. Opin. Ther. Pat. 2000, 10, 575–600. [Google Scholar] [CrossRef]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 717–724. [Google Scholar] [CrossRef]
- Mincione, F.; Scozzafava, A.; Supuran, C.T. The development of topically acting carbonic anhydrase inhibitors as antiglaucoma agents. Curr. Pharm. Des. 2008, 14, 649–654. [Google Scholar] [CrossRef]
- Mack, E.T.; Snyder, P.W.; Perez-Castillejos, R.; Bilgiçer, B.A.; Moustakas, D.T.; Butte, M.J.; Whitesides, G.M. Dependence of avidity on linker length for a bivalent ligand–bivalent receptor model system. J. Am. Chem. Soc. 2012, 134, 333–345. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynninen, P.; Parkkila, S.; Huhtala, H.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Tomas, E. Carbonic anhydrase isozymes II, IX, and XII in uterine tumors. Apmis 2012, 120, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, A.J.; Kivelä, J.; Saarnio, J.; Parkkila, S. Carbonic anhydrases in normal gastrointestinal tract and gastrointestinal tumours. WJG 2005, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Hilvo, M.; Scozzafava, A.; Parkkila, S.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of the new membrane-associated isoform XV with phenols. Bioorg. Med. Chem. Lett. 2008, 18, 3593–3596. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Faizan, M.I.; Dar, S.H.; Ahmad, T.; Rahisuddin. Design and Synthesis of Carbothioamide/Carboxamide-Based Pyrazoline Analogs as Potential Anticancer Agents: Apoptosis, Molecular Docking, ADME Assay, and DNA Binding Studies. ACS Omega 2022, 7, 22639–22656. [Google Scholar] [CrossRef]
- Qian, H.; Chen, W.; Zhang, X.; Zhang, H.; Zhou, J.; Huang, W.; Jin, J.; Dai, D. Synthesis and Biological Evaluation of Dihydroisoquinoline-2 (1H)-Carbothioamide Derivatives as TRPV1 Antagonists. Lett. Drug Des. Discov. 2010, 7, 235–237. [Google Scholar] [CrossRef]
- Afzal, S.; Al-Rashida, M.; Hameed, A.; Pelletier, J.; Sévigny, J.; Iqbal, J. Functionalized Oxoindolin Hydrazine Carbothioamide Derivatives as Highly Potent Inhibitors of Nucleoside Triphosphate Diphosphohydrolases. Front. Pharmacol. 2020, 11, 585876. [Google Scholar] [CrossRef]
- Ali, F.; Shamim, S.; Lateef, M.; Khan, K.M.; Taha, M.; Salar, U.; Wadood, A.; Rehman, A.U.; Nawaz, N.U.A.; Perveen, S. N-Aryl-3, 4-dihydroisoquinoline carbothioamide analogues as potential urease inhibitors. ACS Omega 2021, 6, 15794–15803. [Google Scholar] [CrossRef]
- Çapan, İ.; Servi, S.; Yıldırım, İ.; Sert, Y. Synthesis, DFT Study, Molecular Docking and Drug-Likeness Analysis of the New Hydrazine-1-Carbothioamide, Triazole and Thiadiazole Derivatives: Potential Inhibitors of HSP90. Chem. Select. 2021, 6, 5838–5846. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, X.; Yu, T.; Liu, Y.; Zhang, X.; Yao, Y.; Feng, X.; Liu, H.; Yu, D.; Ma, L.; et al. Discovery of thiosemicarbazone derivatives as effective New Delhi metallo-β-lactamase-1 (NDM-1) inhibitors against NDM-1 producing clinical isolates. APSB 2021, 11, 203–221. [Google Scholar] [CrossRef]
- Arshad, N.; Channar, P.A.; Saeed, A.; Farooqi, S.I.; Javeed, A.; Larik, F.A.; Abbasi, W.A.; Flörke, U. Structure elucidation, DNA binding, DFT, molecular docking and cytotoxic activity studies on novel single crystal (E)-1-(2-fluorobenzylidene) thiosemicarbazide. J. Saudi Chem. Soc. 2018, 22, 1003–1013. [Google Scholar] [CrossRef]
- Malterud, K.E.; Rydland, K.M. Inhibitors of 15-lipoxygenase from orange peel. J. Agric. Food Chem. 2000, 48, 5576–5580. [Google Scholar] [CrossRef] [PubMed]
- Zaib, S.; Saeed, A.; Stolte, K.; Flörke, U.; Shahid, M.; Iqbal, J. New aminobenzenesulfonamide–thiourea conjugates: Synthesis and carbonic anhydrase inhibition and docking studies. Eur. J. Med. Chem. 2014, 78, 140–150. [Google Scholar] [CrossRef]
- Ikai, A. Local rigidity of a protein molecule. Biophys. Chem. 2005, 116, 187–191. [Google Scholar] [CrossRef]
- Sadeghian, H.; Seyedi, S.M.; Saberi, M.R.; Arghiani, Z.; Riazi, M. Design and synthesis of eugenol derivatives, as potent 15-lipoxygenase inhibitors. Bioorg. Med. Chem. 2008, 16, 890–901. [Google Scholar] [CrossRef]
- Ter Haar, D. Foundations of statistical mechanics. RMP 1955, 27, 289–298. [Google Scholar] [CrossRef]
- Jarvis, T.R.; Chughtai, B.; Kaplan, S.A. Testosterone and benign prostatic hyperplasia. Asian J. Androl. 2015, 17, 212. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Khan, A.; Saleem, S.; Idrees, M.; Ali, S.S.; Junaid, M.; Kaushik, A.C.; Wei, D.-Q. Allosteric ligands for the pharmacologically important Flavivirus target (NS5) from ZINC database based on pharmacophoric points, free energy calculations and dynamics correlation. J. Mol. Graph. Model. 2018, 82, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Khan, M.T.; Saleem, S.; Junaid, M.; Ali, A.; Ali, S.S.; Khan, M.; Wei, D.-Q. Structural insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput. Struct. Biotechnoll. 2020, 18, 2174–2184. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | b-CA II | 15-LOX |
|---|---|---|
| (IC50 (μM) ± SEM) a | ||
| 3a | 0.31 ± 0.01 | 0.37 ± 0.02 |
| 3b | 4.97 ± 0.16 | ---b |
| 3c | 2.02 ± 0.12 | 0.16 ± 0.01 |
| 3d | 0.28 ± 0.01 | ---b |
| 3e | 0.38 ± 0.02 | 1.34 ± 0.14 |
| 3f | 10.23 ± 0.21 | 0.82 ± 0.19 |
| 3g | 0.84 ± 0.05 | ---b |
| 3h | 0.13 ± 0.01 | 0.14 ± 0.01 |
| 3i | 0.58 ± 0.01 | 0.54 ± 0.02 |
| 3j | 3.71 ± 0.25 | ---b |
| Acetazolamide | 0.96 ± 0.18 | ---b |
| Quercetin | 15.8 ± 0.61 | |
| Complexes | MM-GBSA (kcal/mol) | ||
|---|---|---|---|
| vdW | Electrostatic | Total Binding Energy | |
| 3h-15-LIPOX | −44.51 | −19.22 | −57.84 |
| 3h-b-CA II | −46.38 | −18.64 | −53.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Channar, P.A.; Alharthy, R.D.; Ejaz, S.A.; Saeed, A.; Iqbal, J. Synthesis, Biological Evaluation, and Molecular Dynamics of Carbothioamides Derivatives as Carbonic Anhydrase II and 15-Lipoxygenase Inhibitors. Molecules 2022, 27, 8723. https://doi.org/10.3390/molecules27248723
Channar PA, Alharthy RD, Ejaz SA, Saeed A, Iqbal J. Synthesis, Biological Evaluation, and Molecular Dynamics of Carbothioamides Derivatives as Carbonic Anhydrase II and 15-Lipoxygenase Inhibitors. Molecules. 2022; 27(24):8723. https://doi.org/10.3390/molecules27248723
Chicago/Turabian StyleChannar, Pervaiz Ali, Rima D. Alharthy, Syeda Abida Ejaz, Aamer Saeed, and Jamshed Iqbal. 2022. "Synthesis, Biological Evaluation, and Molecular Dynamics of Carbothioamides Derivatives as Carbonic Anhydrase II and 15-Lipoxygenase Inhibitors" Molecules 27, no. 24: 8723. https://doi.org/10.3390/molecules27248723