Abstract
Current therapy against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) are based on the use of Remdesivir 1, Molnupiravir 2, and the recently identified Nirmatrelvir 3. Unfortunately, these three drugs showed some limitations regarding potency and possible drug–drug interactions. A series of derivatives coming from a decoration approach of the privileged scaffold s-triazines were synthesized and evaluated against SAR-CoV-2. One derivative emerged as the hit of the series for its micromolar antiviral activity and low cytotoxicity. Mode of action and pharmacokinetic in vitro preliminary studies further confirm the role as candidates for a future optimization campaign of the most active derivative identified with this work.
1. Introduction
Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) is a positive-strand RNA virus [1], belonging to the family of Corona viridae, discovered in late 2019 [2]. It is the causative agent of the coronavirus disease 2019 (COVID-19), that led to the pandemic outbreak and the consequent global lock-down measures of virus containment in 2020. Severe diseases in animals and in humans are associated with COVID-19 at gastrointestinal and, more importantly, respiratory level, with severe pneumonia, high fever, dry cough, and difficulty in breathing as common symptoms at the onset of illness [3]. Worldwide, vaccination has been the main method to reduce the spread of infection and the severity of disease in healthy individuals. In SARS-CoV-2 infected patients at high risk of disease progression, different therapeutic agents targeting directly the viral replication cycle are currently available [4,5], including different monoclonal antibodies, antiviral drugs repurposed, such as Remdesivir 1 (FDA approved) and Molnupiravir 2 (authorized under emergency use authorization, EUA), or recently identified, such as Nirmatrelvir 3 (EUA) (Figure 1). However, the development of novel drugs remains a key priority because currently available antivirals are not highly effective, and their use is limited by a number of drug–drug interactions (Nirmatrelvir), by inconvenient administration (monoclonal antibodies and Remdesivir) or by the toxicity in fragile population (Molnupiravir) [6].
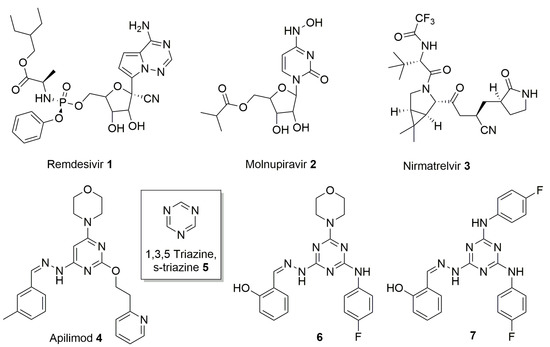
Figure 1.
Chemical structures of Remdesivir 1, Molnupiravir 2, Nirmatrelvir 3, Apilimod 4, s-triazine 5, and two highly decorated triazines previously identified 6 and 7.
1,3,5-Triazine 5, known as symmetric triazine (s-triazine), is an extensively studied heterocyclic nucleus in medicinal chemistry for its easy chemical manipulation and its wide therapeutic application (Figure 1) [7]. s-Triazine is considered, in fact, a privileged structure [8] since it is the core structure of antibacterial, antiviral, anticancer, and antifungal agents [9].
In the context of s-triazine-based compounds, in 2011, our group reported the synthesis of a series of derivatives presenting this privileged core structure, as inhibitor of the human DEAD-box RNA helicase DDX3X, endowed with anti-HIV activity [10]. DDX3X is a potential target for the development of anticancer and antiviral compounds considering its oncogenic role in promoting cancer progression and its active involvement in the replication of different viruses [11,12,13]. From the library of compounds synthesized, two members (compounds 6 and 7, Figure 1) emerged for their antiretroviral effect in the low-micromolar range (EC50 = 2.5 and 2.0 μM, respectively), even if accompanied by a moderate cytotoxicity (CC50 = 10.0 and 8.0 μM, respectively) in peripheral blood mononuclear cells (PBMC).
Driven by our interest in the functionalization of privileged scaffolds [14] and by the possibility of developing novel antiviral agents, we decided to obtain and to test s-triazine-based compounds, starting from 6 and 7, against the emerging SARS-CoV-2. Herein, is described the synthesis and the biological evaluation of a focused library of nine highly decorated triazines as inhibitor of the above-mentioned coronavirus. In particular, substituents with precise functions have been introduced on the azine core to investigate the chemical space around the privileged nucleus. Salicyl aldehyde, (Scheme 1, compound 11a) and 4-fluoro aniline were chosen due to the high antiretroviral activity showed by derivatives 6 and 7. In addition, 4-acetamidobenzaldehyde (Scheme 1, compound 11b) was selected on the basis of the inhibitory activity on DDX3X of some 4-acetamido-phenyl-triazines presented in our previous work [10]. Morpholine was chosen on the basis of the activity of 6 and 7 and also for its capacity to improve drug-like and pharmacokinetic properties [15]. Finally, 3-tolualdehyde (Scheme 1, compound 11c) was used to reproduce a pharmacophoric moiety present on Apilimod (Figure 1, compound 4), a kinase inhibitor repurposed toward SARS-CoV-2 [16] and structurally similar to the hit compounds 6 and 7, already identified. In addition, in vitro absorption, distribution, metabolism, and excretion (ADME) tests were conducted on the most active compound to further confirm its positive physical chemical properties and its potential role as optimizable hit compound.
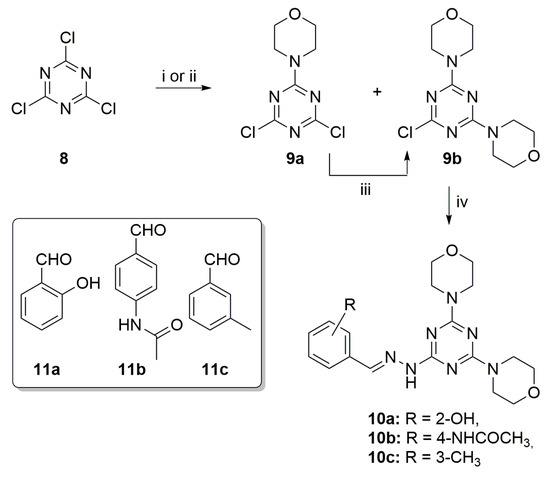
Scheme 1.
Synthesis of triazine derivatives 10a–c. Reagents and conditions: (i) DME, −60 °C, morpholine, 5 h; (ii) N,N-Diisopropylethylamine (DIPEA), morpholine, EtOH, 25 °C, 18 h; (iii) CH2Cl2, 25 °C, morpholine, 18 h; (iv) 1. CH2Cl2, NH2NH2, reflux 12 h, 2. toluene, 11a–c, 3 h, reflux, Dean–Stark.
2. Results and Discussion
A series of three highly decorated triazine derivatives 10a–c, was synthesized starting from 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) 8 following the synthetic procedure reported in Scheme 1.
Compound 8 (1.0 mmol) was reacted with morpholine (1.0 mmol) at −60 °C in dimethoxymethane as solvent, affording a mixture of the monosubstituted 9a and disubstituted 9b intermediates, isolated after flash column chromatography with a 55% and 15% yield, respectively. Attempts to obtain only compound 9a, such as decreasing the temperature to −78 °C or the use of a lower amount of morpholine (0.7 mmol instead of 1.0 mmol) failed. On the other hand, it was possible to directly afford compound 9b using a higher amount (2.2 mmol) of morpholine at 25 °C or reacting the latter (2.0 mmol) with 9a in CH2Cl2 at 25 °C. Subsequently, from the disubstituted intermediate 9b, the three final compounds 10a–c can be synthesized by displacement of the third chlorine atom with hydrazine, and reductive amination with the desired aromatic aldehydes 11a–c to form an imine linkage with a 50, 25, and 45% yield, respectively.
As depicted in Scheme 2, a closely related pathway was used to synthesize compounds 6 and 7 and the highly decorated triazines 13a–b and 14a–b. The reaction of 8 (1.0 mmol) with 4-fluoroaniline (1.0 mmol) at −60 °C or even at −78 °C furnished unselectively compounds 12a and 12b with a 47% and 10% yield, after chromatographic purification. The monosubstituted 12a can be easily converted in the disubstituted 12b by reaction with 4-fluoroaniline and N,N-Diisopropylethylamine (DIPEA) at 25 °C with a 74% yield. Then, 12b is converted into the final derivatives 7, 13a–b by nucleophilic substitution in the presence of hydrated hydrazine and reaction with the opportune aldehyde.
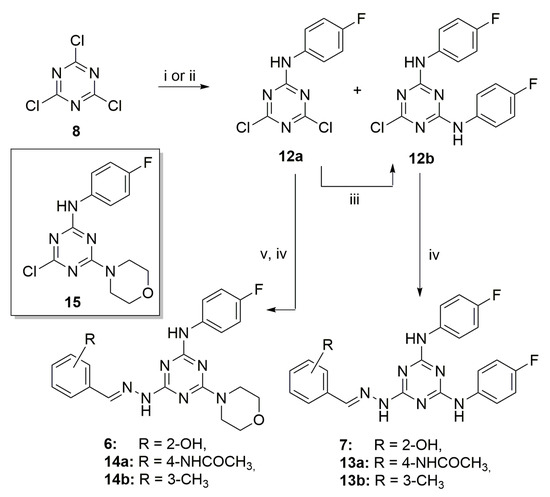
Scheme 2.
Synthesis of triazine derivatives 6, 7, 13a–b and 14a–b. Reagents and conditions: (i) DME, −60 °C, 4-fluoroaniline, 5 h; (ii) DIPEA, 4-fluoroaniline, EtOH, 25 °C, 18 h; (iii) CH2Cl2, 25 °C, 4-fluoroaniline, DIPEA 18 h; (iv) 1. CH2Cl2, NH2NH2, reflux 12 h, 2. MeOH, 11a–c, acetic acid, 25 °C, 18 h; (v) CH2Cl2, 25 °C, morpholine, 5 h.
Finally, the monosubstituted derivative 12a was subjected to aromatic nucleophilic substitution with morpholine to give compound 15 (Scheme 2). From intermediate 15, the trisubstituted triazines 6, 14a–b were synthesized by chlorine displacement with hydrated hydrazine and the addition, in turn, of salicyl aldehyde, 4-acetamidobenzaldehyde, and 3-tolualdehyde 11a–c, respectively. Full details of the synthetic procedures and characterization data are included in the Supplementary Materials.
The inhibitory activity of the resynthesized compounds and the novel triazines designed against SARS-CoV-2 was analyzed in vitro, using the marketed drug Remdesivir as reference (Table 1). The human colon epithelial carcinoma cell line Caco-2, permissive for SARS-CoV-2 infection [17], was used to determine the cytotoxicity and the anti-SARS-CoV-2 activity of compounds 6, 7, 10a–c, 13a,b, and 14a,b as previously described [18] with minor modifications as indicated in the Materials and Methods section below. Briefly, the semi confluent Caco-2 cell line was infected with SARS-CoV-2 at 0.004 Multiplicity of Infection (MOI) in the presence of serial dilutions of the investigational and reference compound (Remdesivir), starting from the non-toxic dose, as previously determined in cytotoxic assay. After 72 h incubation, the antiviral activity of the compounds was measured by immunodetection of the viral N protein in the cell monolayer and expressed as half-maximal inhibitory concentration (IC50). The Selectivity Index (SI) of compounds was calculated as the ratio between the half-maximal cytotoxic concentration (CC50) and the IC50.

Table 1.
Antiviral activity, cytotoxicity, and selectivity index of the synthesized compounds in vitro in a cell-based model 1.
Six compounds, namely 10a–c, 13a,b, and 14a showed a low cytotoxicity (Table 1, entries 1–3, 5, 6, and 8, respectively), compared to the derivatives obtained in the previous study, derivatives 6 and 7 (Table 1, entries 4 and 7). These results highlighted the importance of the introduction of other pharmacophoric moieties, namely the 3-tolyl and 4-acetamidophenyl portions, compared to the salicylic one, as well as the presence of two morpholine substituents on the azine core structure. Regarding the antiviral effect, compound 10a is the only one that highlighted micromolar (12.1 μM) activity against SARS-CoV-2. Even if the activity of 10a was lower than that reported for Remdesivir, the low cytotoxicity showed by this compound prompted us to further evaluate its pharmacokinetic properties, in the light of possible future chemical optimizations.
In addition, all the novel compounds synthesized were also evaluated for their antiretroviral activity but, surprisingly, they seem to be unable to inhibit HIV replication compared to parent compounds 6 and 7 (Table S1 (Supplementary Materials)).
For the more promising compound, derivative 10a, the inhibition of the ATPase activity of the human helicase DDX3X was evaluated. Different concentrations of the compound were incubated with 1 µM of purified recombinant DDX3X and ATPase activity was determined with the ADP-glo assay. This assay showed that the highly decorated triazine 10a is able to inhibit DDX3X with a 6.6 μM ID50 value (Figure S1). This result further confirms the association of the antiviral activity with the inhibition of the human helicase already observed for the parent compounds 6 and 7.
In order to perform the structure–activity relationship (SAR) studies, the in vitro ADME profile of compounds 6, 7, 10a–c (Figure 2) was intensively investigated.
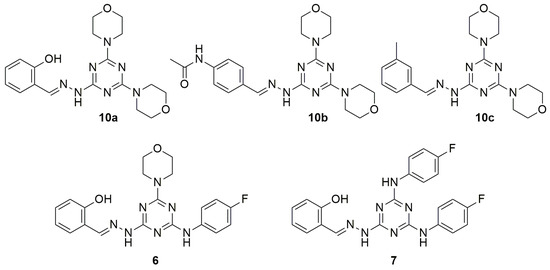
Figure 2.
Structure of compounds 6, 7, 10a–c.
Firstly, the water solubility of compounds was studied dissolving 1 mg of solid compound into 1.0 mL of Mill-Q H2O. As reported in Table 2, the results obtained suggested a good water solubility for compounds 10a–c, with LogS values around −5. The replacement of one or two morpholine moieties with para fluorophenyl groups, worsens the solubility of compounds 6 and 7, to less than 0.1 µg/mL (limit of detection, LOD).
Table 2.
Water solubility of compounds 6, 7, 10a–c.
The trend appreciated in terms of solubility was confirmed by results obtained from permeability studies. In fact, parallel artificial membrane permeability assay (PAMPA) underlined how the low solubility of compounds 6 and 7 was accompanied by a major tendency of these derivatives to interact with the lipidic artificial membrane where they probably remained entrapped. Hence, compounds 6 and 7 resulted in being characterized by Papp of 4.74 and 4.58 cm/s × 10−6 and a general high percentage of membrane retention (MR 28.5% and 15.5%, respectively). Compounds 10a–c, endowed with a higher water solubility, showed a lower tendency to interact with the lipidic bilayer (MR < 10%) and an improved capability to cross the membrane for passive diffusion (Papp from 7.89 to 9.78 cm/s × 10−6) (Table 3).
Table 3.
In vitro PAMPA permeability studies of compounds 6, 7, 10a–c.
From a metabolic point of view, all compounds resulted in a very high stability (more than 99.9%) when incubated for 1 h in the presence of human liver microsomes (HLM). Finally, further studies were performed in order to investigate the stability in the presence of human plasma. All compounds were incubated at the fixed concentration of 100 µM in plasma for 24 h. As reported in Table 4, all compounds resulted in not being affected by the hydrolytic action of plasma esterase with percentages of plasma stability of 98–99% after 24 h of incubation.
Table 4.
Plasma stability studies after 24 h of compounds 6, 7, 10a–c.
The replacement of one or both the 4-fluoroaniline substituents, of compounds 6 and 7, with morpholine increased the water solubility and positively influenced parallel artificial membrane crossing and membrane retention. Compounds 10a–c, in fact, showed better pharmacokinetic properties compared to 6 and 7. In addition, the presence of two morpholines lead to a decrease in the cytotoxicity. Regarding the aromatic hydrazone moiety, salicyl residue of 10a contributed to the most pronounced decrease in the cytotoxicity and to the anti-SARS-CoV-2 activity. The 4-acetamidophenyl and 3-toluic groups of 10b and 10c, respectively, led to better data of membrane permeability but not to antiviral effects.
3. Materials and Methods
3.1. Chemistry-General Part
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. Reagents were obtained from commercial suppliers (Merck Srl, Milan, Italy) and used without further purification. TLC chromatography was performed on precoated aluminum silica gel SIL G/UV254 plates (Macherey-Nagel GmbH & Co. Düren, Germany). The detection occurred via fluorescence quenching or development in a ninhydrin solution (0.2 g of ninhydrin in 99.5 mL ethanol and 0.5 mL acetic acid.). Merck silica gel 60 was used for chromatography (23–400 mesh). 1H NMR and 13C NMR spectra were measured on a Bruker Avance DRX400 (400 MHz/100 MHz) spectrometer. Chemical shifts for protons were reported in parts per million (ppm, δ scale) and internally referenced to the deuterated dimethyl sulfoxide (DMSO-d6), methanol (CD3OD) or chloroform (CDCl3) signal at δ 2.50, 3.33 and 7.28 ppm, respectively. 1H-NMR spectra are reported in this order: multiplicity and number of protons. Signals were characterized as: s (singlet); d (doublet); dd (doublet of doublets); t (triplet); m (multiplet); bs (broad signal). Mass spectra were recorded with an Agilent 1100 LC/MSD VL system (G1946C) (Agilent Technologies, Palo Alto, CA, USA).
3.2. Biology
3.2.1. Cells and Viruses
The SARS-CoV-2 strain belonging to lineage B.1 (EPI_ISL_2472896) was kindly provided by the Department of Biomedical and Clinical Sciences Luigi Sacco, University of Milan (Italy). Once expanded in VERO E6 (African green monkey kidney-cell line, ATCC catalog. N. CRL-1586), the SARS-CoV-2 viral stock was stored at −80 °C and titrated by plaque assay, as previously described [19]. HIV-1 wild-type reference strain NL4-3 (catalog. n. ARP2006), was obtained through the NIH AIDS Reagent Program and viral titer was calculated in TZM- bl cells through the detection of β-galactosidase expression, as previously described [20].
The Caco-2 adherent cell line (ATCC catalog. n. HTB-37), derived from a human colon carcinoma, was used to determine the cytotoxicity and the antiviral activity of candidate compounds against SARS-CoV-2. The H9 suspension cell line (repository code ARP0001; NIBSC Centre for AIDS reagents) derived from a human T cell lymphoma was used to evaluate compounds against HIV-1 in combination with the TZM-bl (repository code ARP5011, NIBSC Centre for AIDS reagents) adherent cell line as described in the antiviral assays’ section.
Adherent cell lines were propagated in high glucose Dulbecco’s Modified Eagle’s Medium with sodium pyruvate and L-glutamine (DMEM; Euroclone), for TZM-bl, or Minimum Essential Medium Eagle (EMEM; Euroclone), for Caco-2, supplemented with 10% Fetal Bovine Serum (FBS; Euroclone) and 1% Penicillin/Streptomycin (Pen/Strep; Euroclone). The propagation medium with a lower concentration of FBS (1%) was used for viral propagation, cytotoxicity, and antiviral activity experiments in adherent cell lines. Suspension cells were propagated in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS; Euroclone), 2 mM L-glutamine, and 1% Penicillin/Streptomycin (Pen/Strep; Euroclone). Cells were incubated at 37 °C in a humidified incubator supplemented with 5% CO2.
3.2.2. Drugs and Cytotoxicity Assay
The cytotoxicity of investigational compounds was determined by CellTiter-Glo 2.0 Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol. Cell viability was calculated by measuring cellular ATP as a marker of metabolically active cells through a luciferase-based chemical reaction. The luminescent signal obtained from cells treated with serial dilution of the investigational compounds, or DMSO as control, was measured through the GloMax® Discover Multimode Microplate Reader (Promega) and elaborated with the GraphPad PRISM software version 9 (La Jolla) to calculate the half-maximal cytotoxic concentration (CC50). Remdesivir (MCE® cat. HY-104077) and Raltegravir (MCE® cat. HY-10353), used as reference compounds for SARS-CoV-2 and HIV-1 antiviral tests, respectively, were purchased from MedChem Express and dissolved in water (Raltegravir) or 100% DMSO (Remdesivir). Following the determination of the CC50, for each compound a non-toxic dose was chosen and used as the starting drug concentration in the subsequent antiviral assays.
3.2.3. Antiviral Assays-SARS-CoV-2
To determine the antiviral activity of candidate compounds against SARS-CoV-2, a direct-yield reduction assay, based on the infection of cells in the presence of serial drug dilutions, was performed as previously described, with minor modifications. Briefly, Caco-2 cells, pre-seeded in a 96-well format, were infected with SARS-CoV-2 viral stock at 0.004 multiplicity of infection (MOI). After 1h adsorption at 37 °C, the viral inoculum was removed and serial dilutions of each tested compound, starting from the non-toxic dose, were added to the infected cells. After 72 h incubation, the antiviral activity was measured in the cell monolayer by immunodetection assay (IA), as previously described [18].
Absorbance was measured at 450 nm optical density (OD450) using the Absorbance Module of the GloMax® Discover Multimode Microplate Reader (Promega). In each plate was included the corresponding reference compound, the mock control (uninfected cells), the virus control, and the virus back titration, performed by diluting 2-fold the initial viral inoculum. Each IA run was validated when both the OD450 values of virus control and the first 2 dilutions of the virus back titration were above 1 OD450. All drug concentrations were tested in duplicate in two independent experiments and in each plate, Remdesivir was used as a reference compound. Infected and uninfected cells without drugs were used to calculate the 100% and 0% of viral replication, respectively. The half-maximal inhibitory concentration (IC50) was calculated through a non-linear regression analysis of the dose–response curves generated with GraphPad PRISM software version 9. The Selectivity Index (SI) of compounds was calculated as a ratio between CC50 and IC50.
3.3. Enzymatic Assay
The ATPase enzymatic activity of DDX3X was determined by the ADP-Glo kit (Promega, MD, USA). The reaction was performed in ATPase buffer (20 mM Tris–HCl pH 8, 2 mM DTT, 70 mM KCl, 2 mM MgCl2, 5% DMSO) pre-incubating 1 µM DDX3X with different concentrations of inhibitor for 10 min at 25 °C. ATPase reaction started with the addition of 1 mM ATP. After 30 min of incubation at 25 °C, the assay was conducted following the manufacturer’s instructions. The ATPase enzymatic activity was analyzed using a 384-wells plate and the GloMax Discover Microplate Reader (Promega, MD, USA) and expressed as a percentage of inhibition calculated from the light units obtained.
3.4. In Vitro ADME
3.4.1. HPLC/UV-MS Method
LC chromatographic analyses were performed by UV/LC-MS with an Agilent 1100 LC/MSD VL system (G1946C) (Agilent Technologies, Palo Alto, CA) equipped with a vacuum solvent degassing unit, a binary high-pressure gradient pump, an 1100 series UV detector, and a 1100 MSD model VL benchtop mass spectrometer. Chromatographic separations were obtained using a Phenomenex Kinetex C18-100 Å column (150 × 4.6 mm) with 5 µm particle size and gradient elution with a binary solution; (eluent A: H2O acidified with formic acid (FA) 0.1% v/v, eluent B: ACN/MeOH 1:1 v/v) at room temperature. The analysis started with 5% of B (from t = 0 to t = 1 min), then B was increased to 95% (from t = 1 to t = 10 min), then kept at 95% (from t = 10 to t = 15 min) and finally returned to 5% of eluent A in one minute. The instrument worked in positive mode and the UV detector operated at 254 nm.
3.4.2. Aqueous Solubility
One mg of each compound was added with 1 mL of Mill-Q H2O. The samples were maintained under shaking at room temperature (RT) overnight. The suspensions were filtered using a 0.45 µm nylon filter (Acrodisc), and the amount of solubilized compound was determined with the HPLC-UV-MS method above reported. The quantification of the solubilized compound was created with the appropriate calibration curve realized with stock solutions in DMSO (0.1–100 µg/mL); the limit of detection (LOD) was quantified at 0.1 µg/mL.
3.4.3. Parallel Artificial Membrane Permeability Assay (PAMPA)
In order to assess the apparent permeability of selected compounds, a stock solution in DMSO of each derivative was prepared at the final concentration of 1 mM. By diluting the stocks 1:1 v/v with phosphate buffer (PBS 25 mM, pH 7.4), donor solutions were made. To mimic the gastrointestinal (GI) phospholipidic bilayer, 10 µL of a 1% w/v dodecane solution of phosphatidylcholine (PC) was used to coat filters. The acceptor solution, made of 1:1 v/v DMSO/PBS, was added to each well (300 µL), while the donor solution (150 µL) was added to each well of the filter plate. The sandwich plates were assembled and incubated for 5 h at room temperature. At the time point, the plates were separated, and the amount of compound passed through the phospholipid bilayer was measured by UV/LC-MS. Finally, apparent permeability (Papp) and membrane retention (MR%) were calculated as previously reported by us [21,22].
3.4.4. Metabolic Stability Assay
A DMSO stock solution of tested compounds was incubated in triplicate in the presence of phosphate buffer (25 mM, pH 7.4), human liver microsomal protein (0.2 mg/mL), and in the presence of an NADPH regenerating system in MgCl2 (48 mM) at a final concentration of 50 µM. The metabolic reaction was conducted for 1 h under shaking at 37 °C and then stopped by adding 1.0 mL of cold acetonitrile (ACN). Centrifuging the reaction mixtures for 10 min at 5000 rpm, the supernatant was separated, dried under nitrogen flow, and finally resuspended in 100 µL of methanol (MeOH). The amount of parent drug and the metabolites were determined as previously described [23].
3.4.5. Plasma Stability Assay
A DMSO stock solution of each compound was incubated in triplicate in the presence of human plasma and HEPES buffer (25 mM, 140 mM NaCl, pH 7.4) at the final concentration of 100 µM at 37 °C under shaking. At the selected time point of 0 and 24 h, 50 µL of the mixture was collected, treated with 1.0 mL of cold ACN, and centrifuged at 5000 rpm for 10 min. The supernatant was collected, and the amount of unmodified compound was quantified with the HLPC-UV/MS method above reported. Calculations of modified compounds were made using time zero as 100% of the unmodified compound.
4. Conclusions
In conclusion, a series of highly decorated triazines was designed and synthesized using the s-triazine privileged scaffold as the core structure. The novel derivatives were evaluated against SARS-CoV-2 resulting in the identification of compound 10a as the hit of the series endowed with a micromolar activity. This compound showed also a very low cytotoxicity against Caco-2 cells, making it even more suitable for further optimizations. Preliminary study regarding the mechanism of action of 10a showed the inhibition of the human DEAD-box RNA helicase DDX3X in the micromolar range as its parent derivatives 6 and 7 obtained in a previous study. Helicase inhibition, in fact, is often correlated with the reduction in the replication of several viruses at different steps such as the nuclear export of the newly synthesized genetic material of the pathogen [8,11,13,22].
In addition, in vitro ADME evaluations highlighted that all compounds tested (6, 7, 10a–c) were characterized by an excellent stability both in the presence of HLM and in plasma. While compound 6 and 7 showed a more lipophilic profile with a major affinity towards lipidic bilayer and lower aqueous solubility, compounds 10a–c demonstrated a satisfactory passive permeability accompanied with low percentages of membrane retention and appreciable water solubility. These additional pharmacokinetic and enzymatic data further confirm the role of the hit compound of the series for 10a and its potentiality as the starting point for future structure optimizations.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27248829/s1, synthetic procedures and full characterization of compounds synthesized (1H-NMR, 13C-NMR, MS); Table S1: Antiviral activity and cytotoxicity of the synthesized compounds in vitro in a cell-based model; Figure S1: Inhibition of the ATPase activity of the human helicase DDX3X.
Author Contributions
Conceptualization, L.B., M.Z. and E.D.; methodology, S.C., I.V. (Ilaria Vicenti), F.P. and M.S.; formal analysis, F.G., I.V. (Ilenia Varasi) and C.L.; data curation, S.C., I.V. (Ilaria Vicenti), F.P. and M.S.; writing—original draft preparation, L.B., M.Z. and E.D.; writing—review and editing, R.S.; funding acquisition, L.B., M.Z., E.D. and G.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministero dell’Istruzione, dell’Università della Ricerca Italiano (MIUR), PRIN 2017, project N. 2017BMK8JR, Title “ORIGINALE CHEMIAE in Antiviral Strategy—Origin and Modernization of Multi-Component Chemistry as a Source of Innovative Broad Spectrum Antiviral Strategy” (L.B., R.S., E.D. and M.Z.); Italian Association of Cancer Research (AIRC), grant IG20762 (G.M.); the National Research Council, Project DSB.AD001.180.002 (G.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The Fondazione Adriano Buzzati-Traverso (Donazione Franca Fois) and the non-profit Trust B Solidale ONLUS association supported this research with a fellowship (C.L.).
Conflicts of Interest
M.Z. reports consultancy for ViiV Healthcare, Gilead Sciences, GlaxoSmithKline, Janssen-Cilag, Theratechnologies, Merck Sharp, and Dohme, and grants for his institution from ViiV Healthcare, Theratechnologies, and Gilead Sciences outside the submitted work. All other authors: no conflicts to declare.
References
- Machhi, J.; Herskovitz, J.; Senan, A.M.; Dutta, D.; Nath, B.; Oleynikov, M.D.; Blomberg, W.R.; Meigs, D.D.; Hasan, M.; Patel, M.; et al. The Natural History, Pathobiology, and Clinical Manifestations of SARS-CoV-2 Infections. J. Neuroimmune. Pharmacol. 2020, 15, 359–386. [Google Scholar] [CrossRef] [PubMed]
- WHO. Situation Reports; World Health Organization: Geneva, Switzerland, 2020; Volume 180. [Google Scholar]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.covid19treatmentguidelines.nih.gov (accessed on 30 October 2022).
- Majumdar, M.; Singh, V.; Misra, T.K.; Roy, D.N. In silico studies on structural inhibition of SARS-CoV-2 main protease Mpro by major secondary metabolites of Andrographis paniculata and Cinchona officinalis. Biologia 2022, 77, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Saravolatz, L.D.; Depcinski, S.; Sharma, M. Molnupiravir and Nirmatrelvir-Ritonavir: Oral COVID Antiviral Drugs. Clin. Infect. Dis. 2022, ciac180. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sheyi, R.; de la Torre, B.G.; El-Faham, A.; Albericio, F. s-Triazine: A Privileged Structure for Drug Discovery and Bioconjugation. Molecules 2021, 26, 864. [Google Scholar] [CrossRef]
- Shah, D.R.; Modh, R.P.; Chikhalia, K.H. Privileged s-triazines: Structure and pharmacological applications. Future Med. Chem. 2014, 6, 463–477. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Triazine as a promising scaffold for its versatile biological behavior. Eur. J. Med. Chem. 2015, 102, 39–57. [Google Scholar] [CrossRef]
- Maga, G.; Falchi, F.; Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Irannejad, H.; Manetti, F.; Garbelli, A.; Samuele, A.; et al. Toward the discovery of novel anti-HIV drugs. Second-generation inhibitors of the cellular ATPase DDX3 with improved anti-HIV activity: Synthesis, structure-activity relationship analysis, cytotoxicity studies, and target validation. ChemMedChem 2011, 6, 1371–1389. [Google Scholar] [CrossRef]
- Brai, A.; Fazi, R.; Tintori, C.; Zamperini, C.; Bugli, F.; Sanguinetti, M.; Stigliano, E.; Esté, J.; Badia, R.; Franco, S.; et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. USA 2016, 113, 5388–5393. [Google Scholar] [CrossRef]
- Secchi, M.; Lodola, C.; Garbelli, A.; Bione, S.; Maga, G. DEAD-Box RNA Helicases DDX3X and DDX5 as Oncogenes or Oncosuppressors: A Network Perspective. Cancers 2022, 14, 3820. [Google Scholar] [CrossRef]
- Brai, A.; Trivisani, C.I.; Poggialini, F.; Pasqualini, C.; Vagaggini, C.; Dreassi, E. DEAD-Box Helicase DDX3X as a Host Target against Emerging Viruses: New Insights for Medicinal Chemical Approaches. J. Med. Chem. 2022, 65, 10195–10216. [Google Scholar] [CrossRef]
- Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Botta, M. Practical one-pot two-step protocol for the microwave-assisted synthesis of highly functionalized rhodanine derivatives. J. Comb. Chem. 2010, 12, 200–205. [Google Scholar] [CrossRef]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a privileged structure: A review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef]
- Baranov, M.V.; Bianchi, F.; van den Bogaart, G. The PIKfyve Inhibitor Apilimod: A Double-Edged Sword against COVID-19. Cells 2020, 10, 30. [Google Scholar] [CrossRef]
- Mautner, L.; Hoyos, M.; Dangel, A.; Berger, C.; Ehrhardt, A.; Baiker, A. Replication kinetics and infectivity of SARS-CoV-2 variants of concern in common cell culture models. Virol. J. 2022, 19, 76. [Google Scholar] [CrossRef]
- Grazia Martina, M.; Vicenti, I.; Bauer, L.; Crespan, E.; Rango, E.; Boccuto, A.; Olivieri, N.; Incerti, M.; Zwaagstra, M.; Allodi, M.; et al. Bithiazole Inhibitors of Phosphatidylinositol 4-Kinase (PI4KIIIβ) as Broad-Spectrum Antivirals Blocking the Replication of SARS-CoV-2, Zika Virus, and Human Rhinoviruses. ChemMedChem 2021, 16, 3548–3552. [Google Scholar] [CrossRef]
- Vicenti, I.; Martina, M.G.; Boccuto, A.; De Angelis, M.; Giavarini, G.; Dragoni, F.; Marchi, S.; Trombetta, C.M.; Crespan, E.; Maga, G.; et al. System-oriented optimization of multi-target 2,6-diaminopurine derivatives: Easily accessible broad-spectrum antivirals active against flaviviruses, influenza virus and SARS-CoV-2. Eur. J. Med. Chem. 2021, 224, 113683. [Google Scholar] [CrossRef]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J. Clin. Lab. Anal. 2018, 32, e22206. [Google Scholar] [CrossRef]
- Tintori, C.; Brai, A.; Dasso Lang, M.C.; Deodato, D.; Greco, A.M.; Bizzarri, B.M.; Cascone, L.; Casian, A.; Zamperini, C.; Dreassi, E.; et al. Development and in Vitro Evaluation of a Microbicide Gel Formulation for a Novel Non-Nucleoside Reverse Transcriptase Inhibitor Belonging to the N-Dihydroalkyloxybenzyloxopyrimidines (N-DABOs) Family. J. Med. Chem. 2016, 59, 2747–2759. [Google Scholar] [CrossRef]
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X inhibitors, an effective way to overcome HIV-1 resistance targeting host proteins. Eur. J. Med. Chem. 2020, 200, 112319. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).