
Time and Angle-Resolved Time-of-Flight Electron Spectroscopy for Functional Materials Science

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
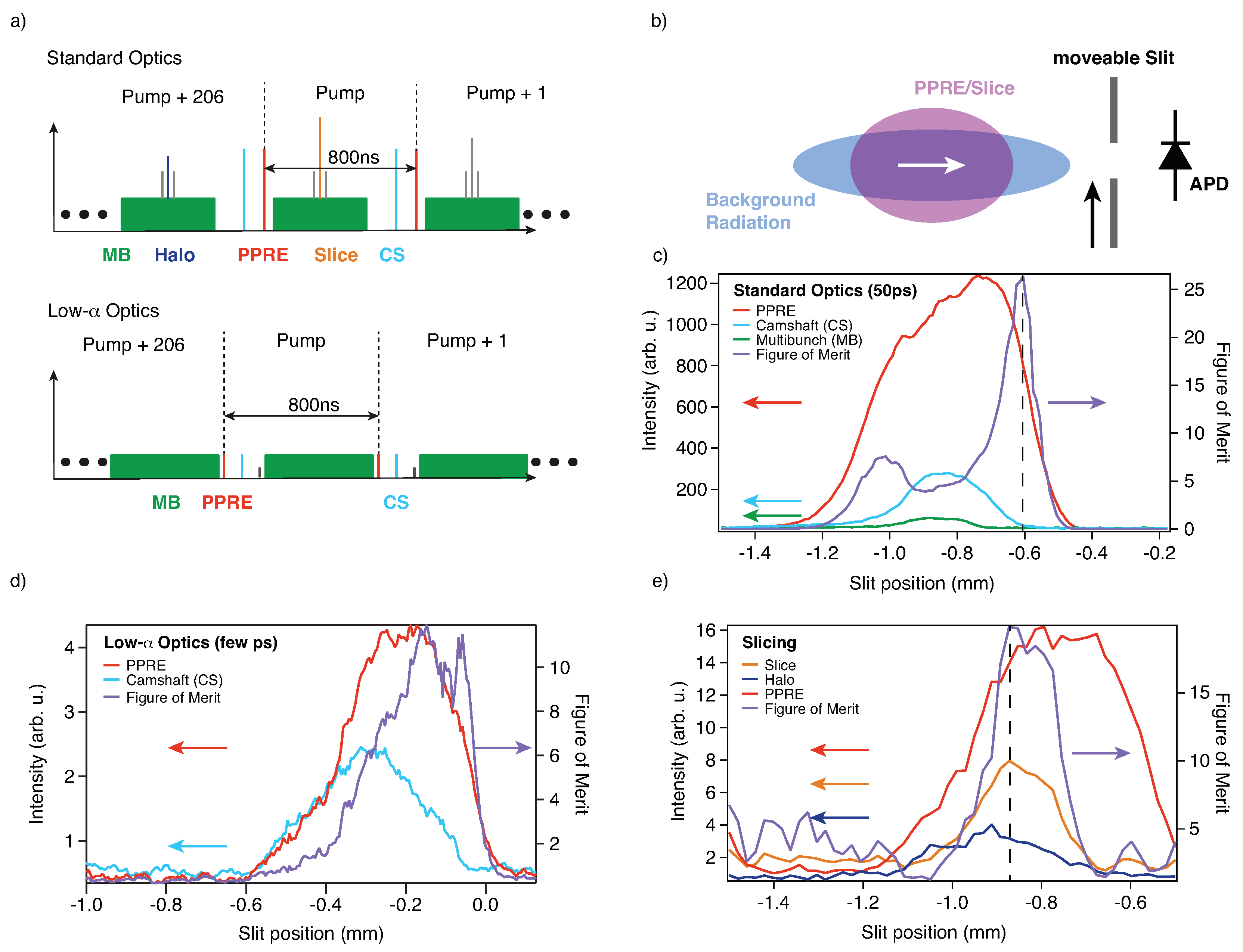
2.1. Extraction of Storage Ring Sub-Patterns
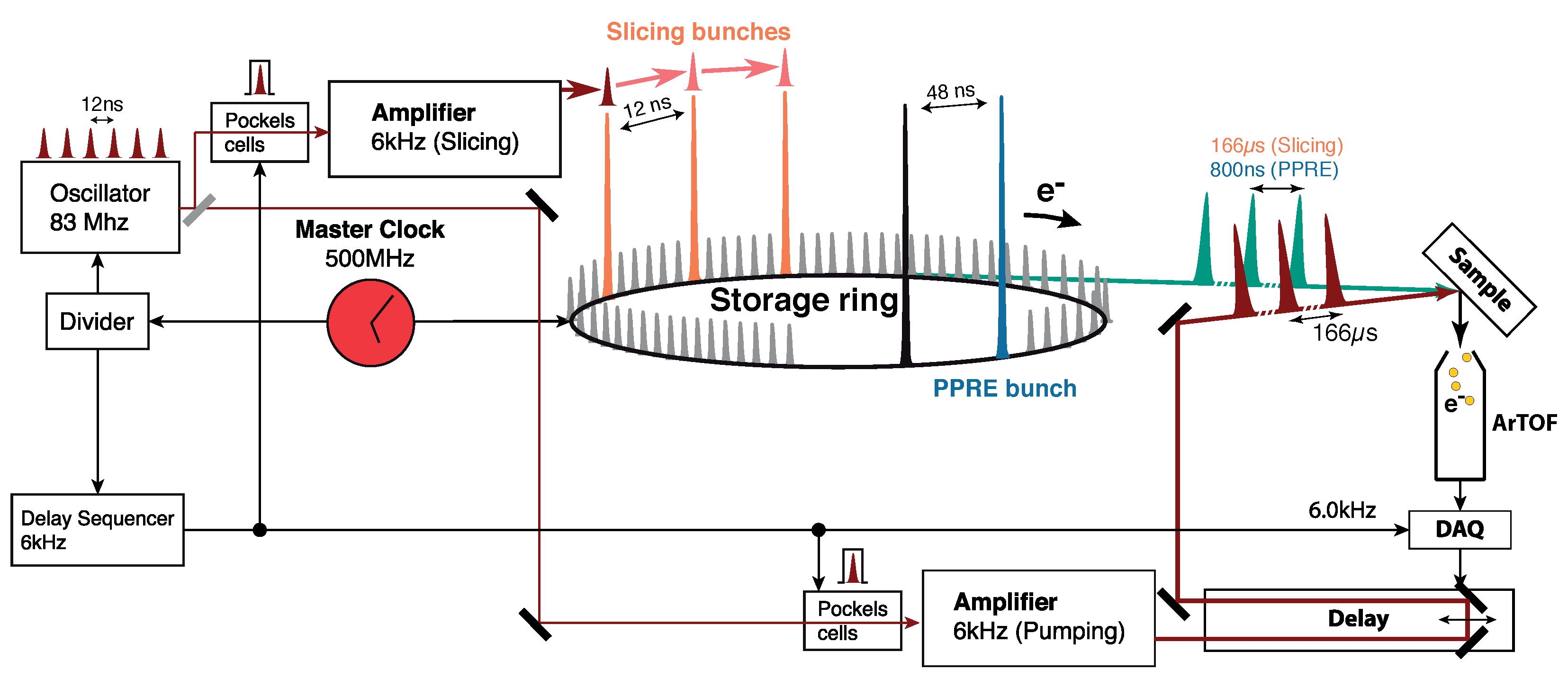
2.2. The Synchronized Optical Laser System for Optical Excitation
2.3. The SurfaceDynamics Instrument
2.4. Measurements Settings and Samples
3. Results and Discussion
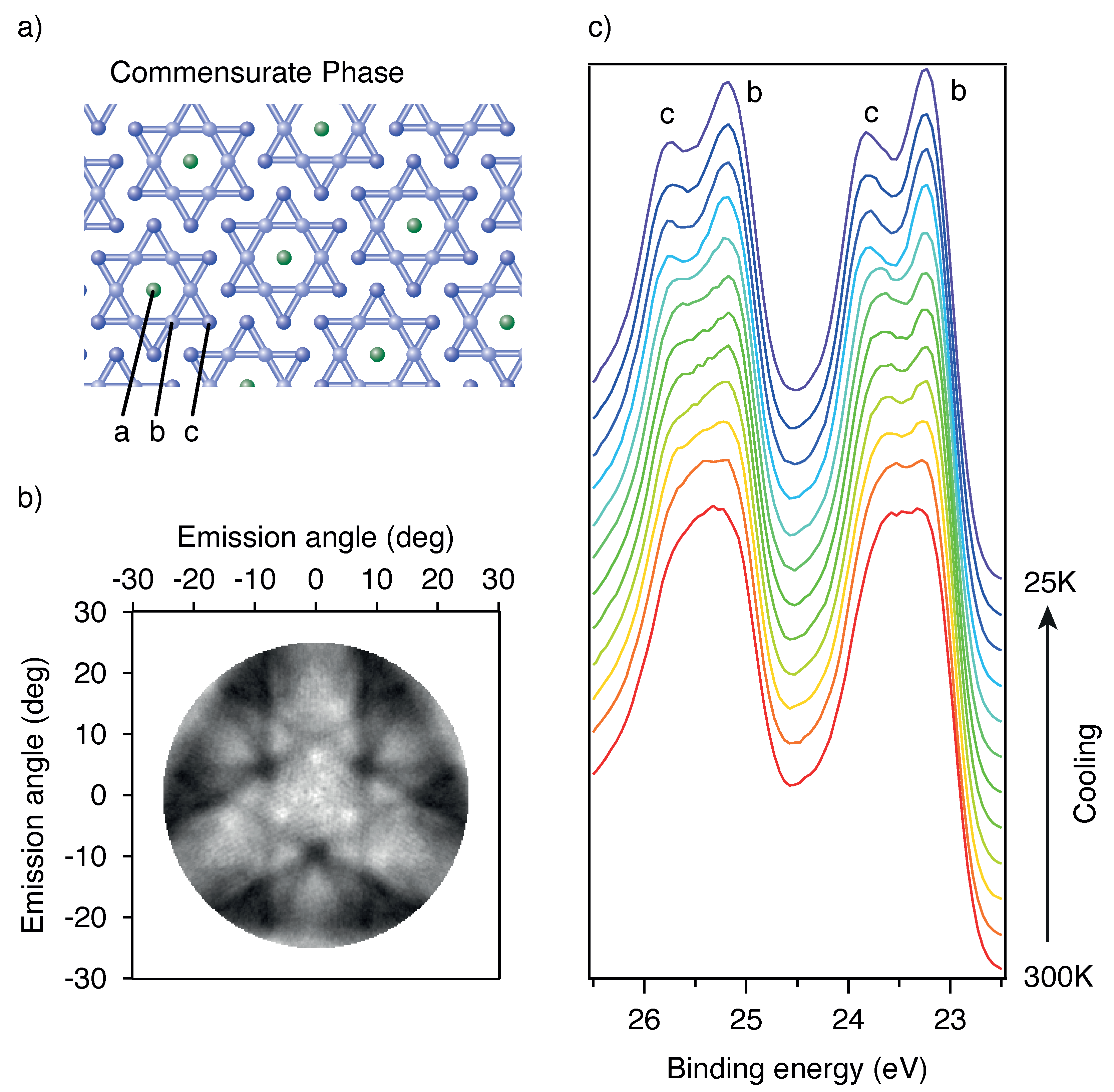
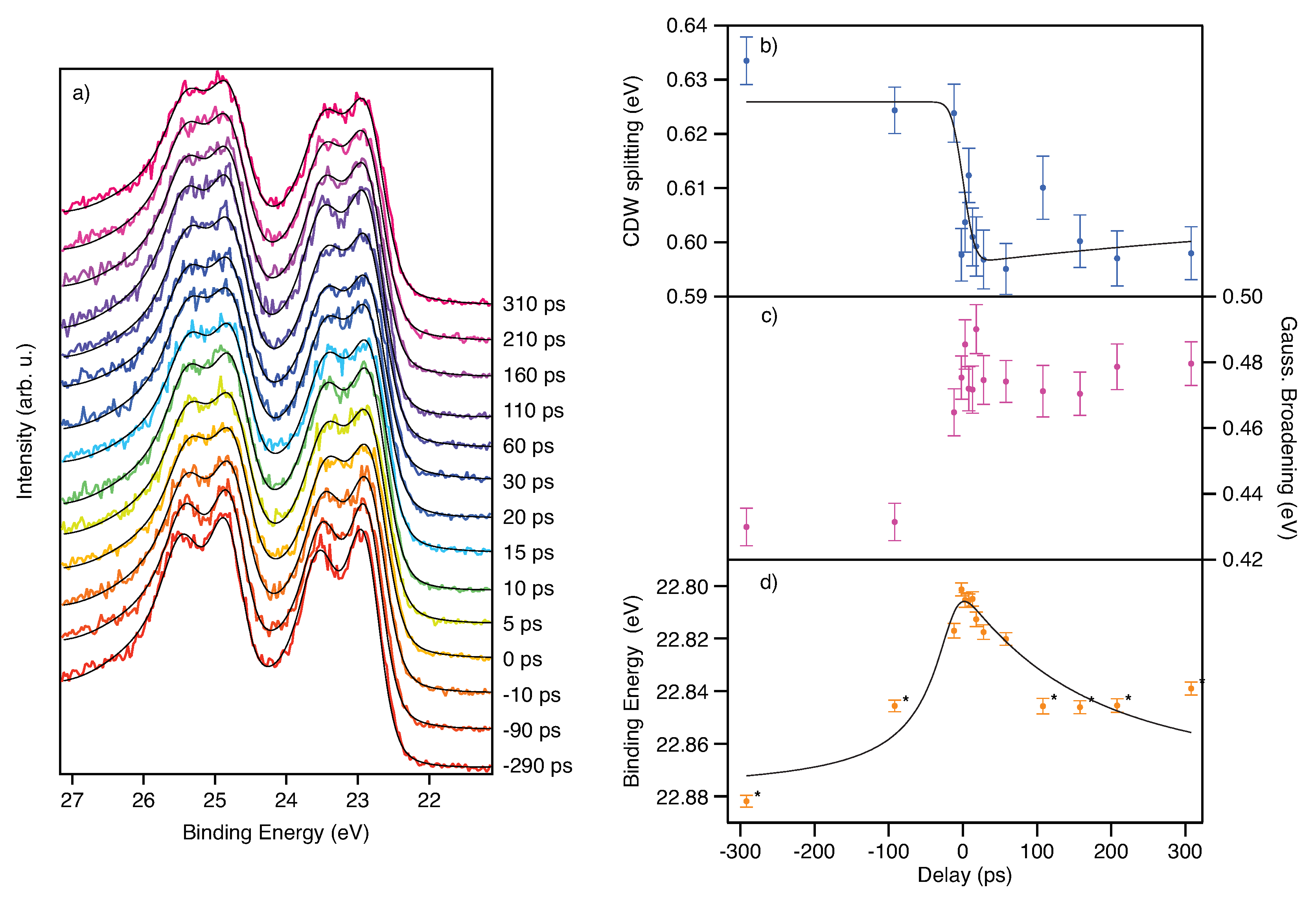
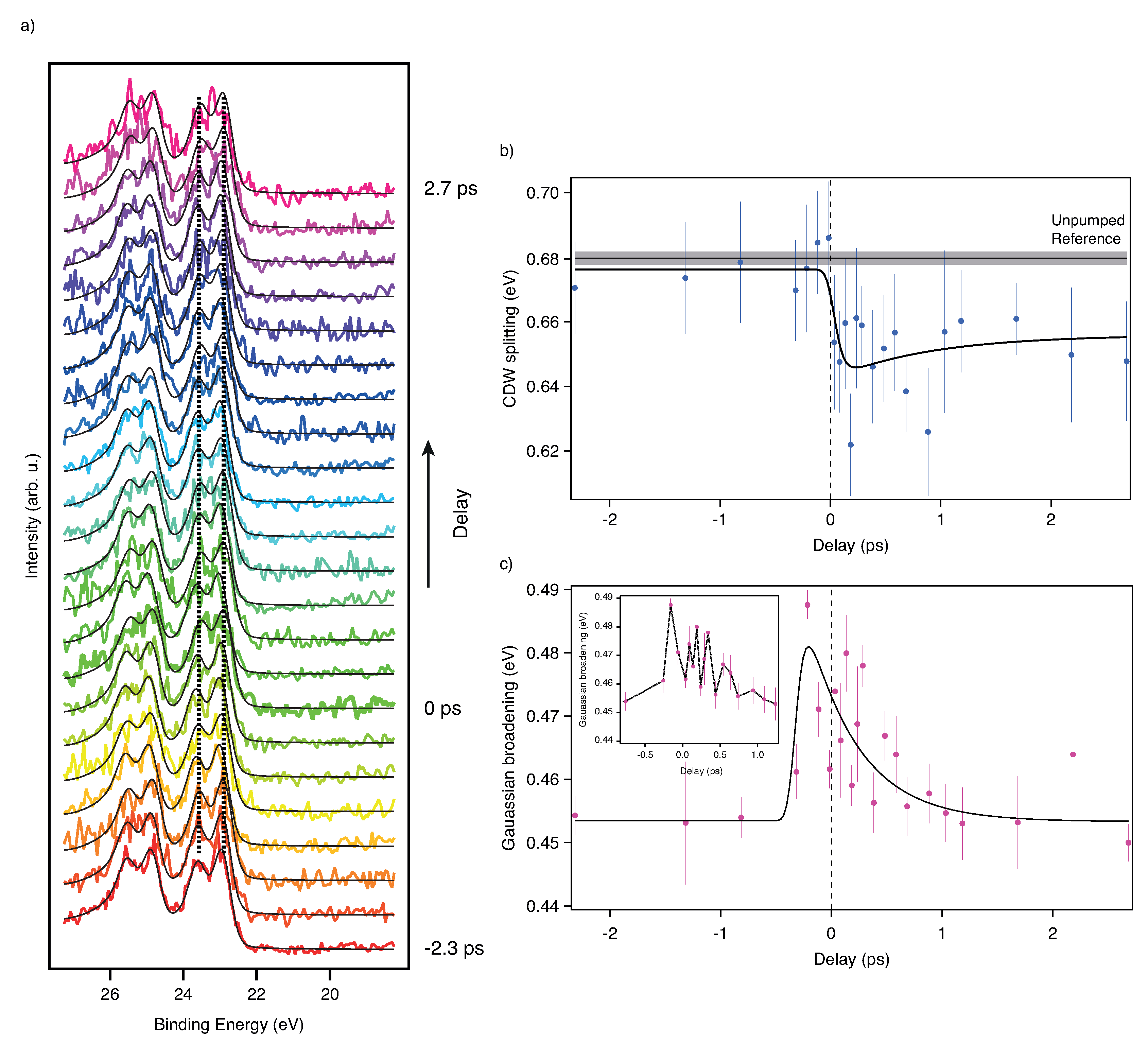
3.1. Time Resolved X-ray Photoelectron Spectroscopy with Different Time Scales at 1T-TaS2
3.2. Towards Time-Resolved X-ray Photoelectron Diffraction at TaS2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ESCA | Electron Spectroscopy for Chemical Analysis |
| XPD | X-ray Photo Electron Diffraction |
| ArTOF | Angle resolved Time-of-Flight |
| FEL | Free-Electron Laser |
| VUV | Vacuum Ultra-Violet |
| SASE | Self-Amplified Spontaneous Emission |
| HHG | Higher Harmonics Generation |
| TaS2 | Tantalumdisulphide |
| PGM | Plane Grating Monochromator |
| PPRE | Pulse Picking by Resonant Excitation |
| TRIBs | Transverse Island Buckets |
| FWHM | Full Width at Half Maximum |
| APD | Avalanche Photo Diode |
| MB | Multibunch |
| CS | Camshaft (bunch) |
| BBO | Beta Barium Borate |
| OPA | Optical Paramteric Amplifier |
| UV | Ultra-Violet |
| ND | Neutral Density |
| AR | Anti Reflective |
| MIM | Minimum Incremental Motion |
| TMP | Turbo Molecular Pump |
| IGP | Ion Getter Pump |
| NEG | Non-Evaporable Getter Pump |
| LHe | Liquid Helium |
| LEED | Low-Energy Electron Diffraction |
| LN2 | Liquid Nitrogen |
| CDW | Charge Density Wave |
| ICCDW | Incommensurate Charge Density Wave |
| NCCDW | Nearly Commensurate Charge Density Wave |
| CCDW | Commensurate Charge Density Wave |
| PLD | Periodic Lattice Distortion |
| XPS | X-ray Photoelectron Spectroscopy |
| SO | Spin-Orbit |
| trPES | Time-Resolved Photoemission Spectroscopy |
| FLASH | Free-Electron Laser in Hamburg |
| HHG | Higher Harmonic Generation |
| SCS | Space-Charge Shift |
References
- Hüfner, S. Photoelectron Spectroscopy: Principles and Applications; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Wannberg, B. Electron optics development for photo-electron spectrometers. Nucl. Instrum. Methods Phys. Res. Sect. Accel. Spectrometers Detect. Assoc. Equip. 2009, 601, 182–194. [Google Scholar] [CrossRef]
- Zhou, X.J.; Wannberg, B.; Yang, W.L.; Brouet, V.; Sun, Z.; Douglas, J.F.; Dessau, D.; Hussain, Z.; Shen, Z.X. Space charge effect and mirror charge effect in photoemission spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2005, 142, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, S.; Sohrt, C.; Beye, M.; Rohwer, T.; Sorgenfrei, F.; Marczynski-Bühlow, M.; Kalläne, M.; Redlin, H.; Hennies, F.; Bauer, M.; et al. Time-resolved X-ray photoelectron spectroscopy at FLASH. New J. Phys. 2012, 14, 013062. [Google Scholar] [CrossRef]
- Öhrwall, G.; Karlsson, P.; Wirde, M.; Lundqvist, M.; Andersson, P.; Ceolin, D.; Wannberg, B.; Kachel, T.; Dürr, H.; Eberhardt, W.; et al. A new energy and angle resolving electron spectrometer–First results. J. Electron Spectrosc. Relat. Phenom. 2011, 183, 125–131. [Google Scholar] [CrossRef]
- Ovsyannikov, R.; Karlsson, P.; Lundqvist, M.; Lupulescu, C.; Eberhardt, W.; Foehlisch, A.; Svensson, S.; Mårtensson, N. Principles and operation of a new type of electron spectrometer—ArTOF. J. Electron Spectrosc. Relat. Phenom. 2013, 191, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Kühn, D.; Sorgenfrei, F.; Giangrisostomi, E.; Jay, R.; Musazay, A.; Ovsyannikov, R.; Stråhlman, C.; Svensson, S.; Mårtensson, N.; Föhlisch, A. Capabilities of Angle Resolved Time of Flight electron spectroscopy with the 60° wide angle acceptance lens. J. Electron Spectrosc. Relat. Phenom. 2018, 224, 45–50. [Google Scholar] [CrossRef]
- Dell’Angela, M.; Anniyev, T.; Beye, M.; Coffee, R.; Föhlisch, A.; Gladh, J.; Kaya, S.; Katayama, T.; Krupin, O.; Nilsson, A.; et al. Vacuum space charge effects in sub-picosecond soft X-ray photoemission on a molecular adsorbate layer. Struct. Dyn. 2015, 2, 025101. [Google Scholar] [CrossRef] [Green Version]
- Holldack, K.; Ovsyannikov, R.; Kuske, P.; Müller, R.; Schälicke, A.; Scheer, M.; Gorgoi, M.; Kühn, D.; Leitner, T.; Svensson, S.; et al. Single bunch X-ray pulses on demand from a multi-bunch synchrotron radiation source. Nat. Commun. 2014, 5, 4010. [Google Scholar] [CrossRef] [Green Version]
- Holldack, K.; Bahrdt, J.; Balzer, A.; Bovensiepen, U.; Brzhezinskaya, M.; Erko, A.; Eschenlohr, A.; Follath, R.; Firsov, A.; Frentrup, W.; et al. FemtoSpeX: A versatile optical pump-soft X-ray probe facility with 100 fs X-ray pulses of variable polarization. J. Synchrotron Rad. 2014, 21, 1090–1140. [Google Scholar] [CrossRef] [Green Version]
- Förster, D.F.; Lindenau, B.; Leyendecker, M.; Janssen, F.; Winkler, C.; Schumann, F.O.; Kirschner, J.; Holldack, K.; Föhlisch, A. Phase-locked MHz pulse selector for X-ray sources. Opt. Lett. 2015, 40, 2265–2268. [Google Scholar] [CrossRef]
- Goslawski, P.; Jankowiak, A.; Kramer, F.; Ries, M.; Ruprecht, M.; Wüstefeld, G. Transverse Resonance Island Buckets as Bunch Separation Scheme. In Proceedings of the 8th International Particle Accelerator Conference (IPAC’17), Copenhagen, Denmark, 14–19 May 2017; JACOW: Geneva, Switzerland, 2017; pp. 3059–3062. [Google Scholar] [CrossRef]
- Jankowiak, A.; Wüstefeld, G. Low-α Operation of BESSY II and Future Plans for an Alternating Bunch Length Scheme BESSYVSR. Synchrotron Radiat. News 2013, 26, 22–24. [Google Scholar] [CrossRef]
- Schick, D.; Le Guyader, L.; Pontius, N.; Radu, I.; Kachel, T.; Mitzner, R.; Zeschke, T.; Schüßler-Langeheine, C.; Föhlisch, A.; Holldack, K. Analysis of the halo background in femtosecond slicing experiments. J. Synchrotron Rad. 2016, 23, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Inada, R.; Onuki, Y.; Tanuma, S. Anderson Localization and Crystalline Defects of 1T-TaS2. J. Phys. Soc. Jpn. 1983, 52, 3536–3543. [Google Scholar] [CrossRef]
- Hellmann, S.; Beye, M.; Sohrt, C.; Rohwer, T.; Sorgenfrei, F.; Redlin, H.; Kalläne, M.; Marczynski-Bühlow, M.; Hennies, F.; Bauer, M.; et al. Ultrafast Melting of a Charge-Density Wave in the Mott Insulator 1T-TaS2. Phys. Rev. Lett. 2010, 105, 187401. [Google Scholar] [CrossRef] [Green Version]
- Rossnagel, K. On the origin of charge-density waves in select layered transition-metal dichalcogenides. J. Physics: Condens. Matter 2011, 23, 213001. [Google Scholar] [CrossRef]
- Sipos, B.; Kusmartseva, A.F.; Akrap, A.; Berger, H.; Forró, L.; Tutiš, E. From Mott state to superconductivity in 1T-TaS2. Nat. Mater. 2008, 7, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Pollak, R.A.; Eastman, D.E.; Himpsel, F.J.; Heimann, P.; Reihl, B. 1T-TaS2 charge-density-wave metal-insulator transition and Fermi-surface modification observed by photoemission. Phys. Rev. B 1981, 24, 7435–7438. [Google Scholar] [CrossRef]
- Stoltz, D.; Stoltz, S.E. X-ray photoelectron diffraction investigation of the cleavage plane in 1T-transition metal dichalcogenides. Phys. B: Condens. Matter 2007, 398, 172–177. [Google Scholar] [CrossRef]
- Ishizaka, K.; Kiss, T.; Yamamoto, T.; Ishida, Y.; Saitoh, T.; Matsunami, M.; Eguchi, R.; Ohtsuki, T.; Kosuge, A.; Kanai, T.; et al. Femtosecond core-level photoemision spectroscopy on 1 T-TaS 2 using a 60-eV laser source. Phys. Rev. B 2011, 83, 081104. [Google Scholar] [CrossRef]
- Hughes, H.P.; Scarfe, J.A. Site specific photohole screening in a charge density wave. Phys. Rev. Lett. 1995, 74, 3069–3072. [Google Scholar] [CrossRef]
- Riffe, D.M.; Hale, W.; Kim, B.; Erskine, J.L. Conduction-electron screening in the bulk and at low-index surfaces of Ta metal. Phys. Rev. B 1995, 51, 11012–11017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, S.; Rohwer, T.; Kalläne, M.; Hanff, K.; Sohrt, C.; Stange, A.; Carr, A.; Murnane, M.M.; Kapteyn, H.C.; Kipp, L.; et al. Time-domain classification of charge-density-wave insulators. Nat. Commun. 2012, 3, 1069. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, S.; Rossnagel, K.; Marczynski-Bühlow, M.; Kipp, L. Vacuum space-charge effects in solid-state photoemission. Phys. Rev. B 2009, 79, 035402. [Google Scholar] [CrossRef]
- Oloff, L.P.; Oura, M.; Rossnagel, K.; Chainani, A.; Matsunami, M.; Eguchi, R.; Kiss, T.; Nakatani, Y.; Yamaguchi, T.; Miyawaki, J.; et al. Time-resolved HAXPES at SACLA: Probe and pump pulse-induced space-charge effects. New J. Phys. 2014, 16, 123045. [Google Scholar] [CrossRef] [Green Version]
- Oloff, L.P.; Chainani, A.; Matsunami, M.; Takahashi, K.; Togashi, T.; Osawa, H.; Hanff, K.; Quer, A.; Matsushita, R.; Shiraishi, R.; et al. Time-resolved HAXPES using a microfocused XFEL beam: From vacuum space-charge effects to intrinsic charge-carrier recombination dynamics. Sci. Rep. 2016, 6, 35087. [Google Scholar] [CrossRef] [PubMed]
- Kühn, D.; Giangrisostomi, E.; Jay, R.M.; Sorgenfrei, F.; Föhlisch, A. The influence of X-ray pulse length on space-charge effects in optical pump/X-ray probe photoemission. New J. Phys. 2019, 21, 073042. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Std. Dev. |
|---|---|---|
| SO Splitting (eV) | 1.912 | 0.001 |
| SO Ratio | 0.846 | 0.002 |
| CDW Ratio | 0.591 | 0.002 |
| Asymmetry site b | 0.289 | 0.001 |
| Asymmetry site c | 0.360 | 0.001 |
| Parameter | Value | Std. Dev. |
|---|---|---|
| SCS Model | ||
| Probe Pulse width (ps) | 29 | 12 |
| Raw Zero Delay (ps) | −8 | 2 |
| # of electrons | 11,100 | 1200 |
| Average kin. Energy (eV) | 11 | 5 |
| Laser Spot Size Diameter (μm) | 400 | fixed |
| CDW Model | ||
| CDW Splitting unpumped (eV) | 0.626 | 0.003 |
| Amplitude fast decay (eV) | 0.030 | 0.005 |
| Amplitude slow recovery (eV) | −0.01 | 0.02 |
| Time constant fast decay (ps) | 0.1 | fixed |
| Time constant slow recovery (ps) | 655 | fixed |
| Parameter | Value | Std. Dev. |
|---|---|---|
| Broadening Dynamics | ||
| Value before t0 (eV) | 0.453 | 0.002 |
| Amplitude fast decay (eV) | −0.043 | 0.006 |
| Amplitude slow recovery (eV) | 0.043 | 0.005 |
| Time constant fast decay (ps) | 0.05 | fixed |
| Time constant slow recovery (ps) | 0.46 | 0.12 |
| t0 position (raw ps) | −210.04 | 0.02 |
| Temporal resolution (ps) | 0.12 | fixed |
| reduced | 3.79 | n.a. |
| CDW Dynamics | ||
| Value before t0 (eV) | 0.676 | 0.007 |
| Amplitude fast Decay (eV) | 0.034 | 0.019 |
| Amplitude slow recovery (eV) | −0.014 | 0.017 |
| Time constant fast decay (ps) | 0.05 | fixed |
| Time constant slow recovery (ps) | 0.9 | 4.2 |
| t0 position (raw ps) | −209.68 | 0.08 |
| Temporal resolution (ps) | 0.12 | fixed |
| reduced | 0.53 | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorgenfrei, N.L.A.N.; Giangrisostomi, E.; Kühn, D.; Ovsyannikov, R.; Föhlisch, A. Time and Angle-Resolved Time-of-Flight Electron Spectroscopy for Functional Materials Science. Molecules 2022, 27, 8833. https://doi.org/10.3390/molecules27248833
Sorgenfrei NLAN, Giangrisostomi E, Kühn D, Ovsyannikov R, Föhlisch A. Time and Angle-Resolved Time-of-Flight Electron Spectroscopy for Functional Materials Science. Molecules. 2022; 27(24):8833. https://doi.org/10.3390/molecules27248833
Chicago/Turabian StyleSorgenfrei, Nomi Lucia Ada Nathalie, Erika Giangrisostomi, Danilo Kühn, Ruslan Ovsyannikov, and Alexander Föhlisch. 2022. "Time and Angle-Resolved Time-of-Flight Electron Spectroscopy for Functional Materials Science" Molecules 27, no. 24: 8833. https://doi.org/10.3390/molecules27248833
APA StyleSorgenfrei, N. L. A. N., Giangrisostomi, E., Kühn, D., Ovsyannikov, R., & Föhlisch, A. (2022). Time and Angle-Resolved Time-of-Flight Electron Spectroscopy for Functional Materials Science. Molecules, 27(24), 8833. https://doi.org/10.3390/molecules27248833