
Cell-Penetrating Peptide–Peptide Nucleic Acid Conjugates as a Tool for Protein Functional Elucidation in the Native Bacterium

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Gene Annotation and Identification of Carrageenase Encoding Genes
2.4. Carrageenan Degradation by Paenibacillus sp. YYML68
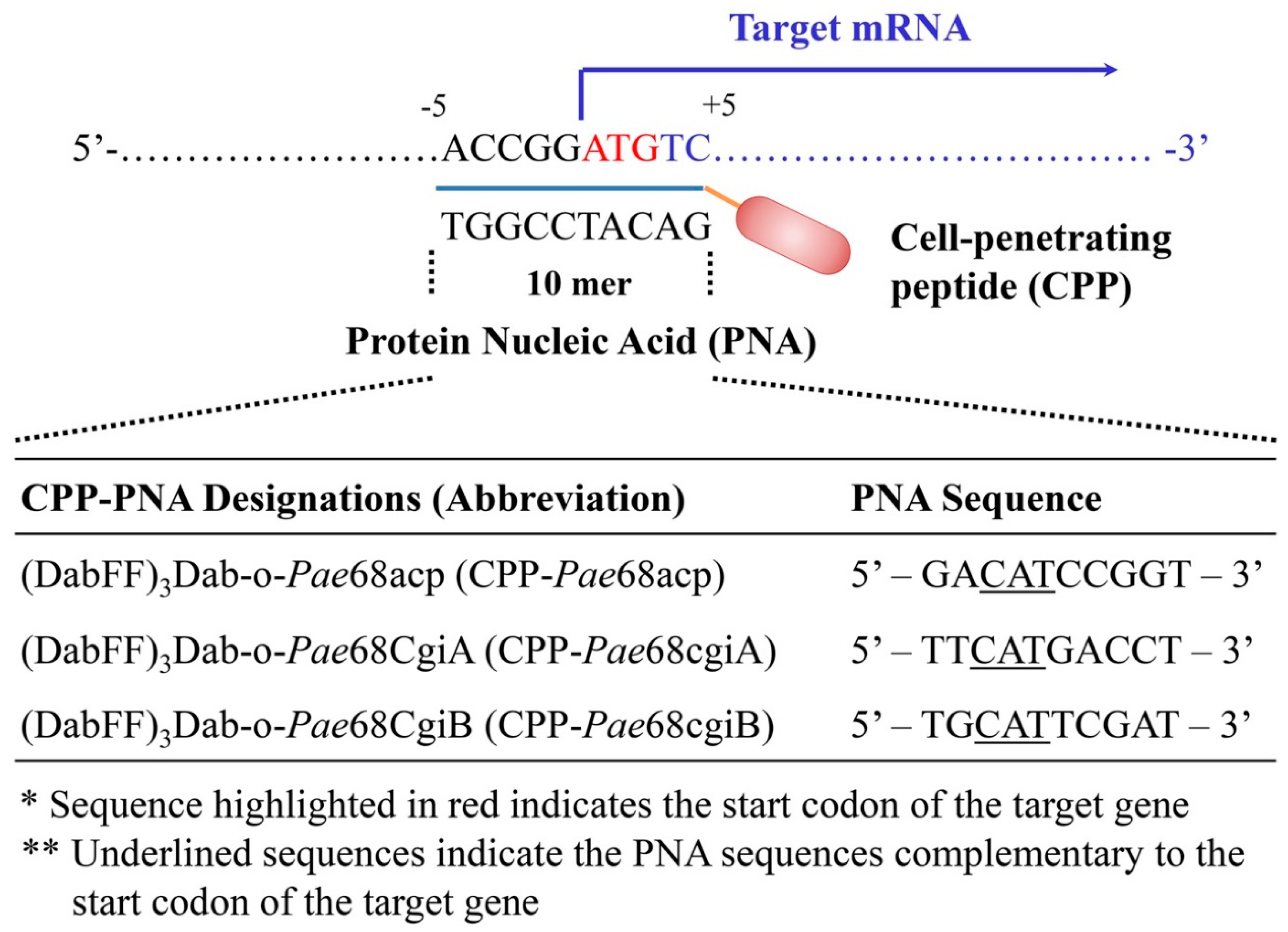
2.4.1. CPP–PNA Design and Synthesis
2.4.2. Inhibition Assays Using CPP–PNA
2.4.3. Protein Structure Prediction and Comparative Analysis
2.5. Graph Presentation and Analysis
3. Results
3.1. Candidate Carrageenase Genes in Paenibacillus sp. YYML68
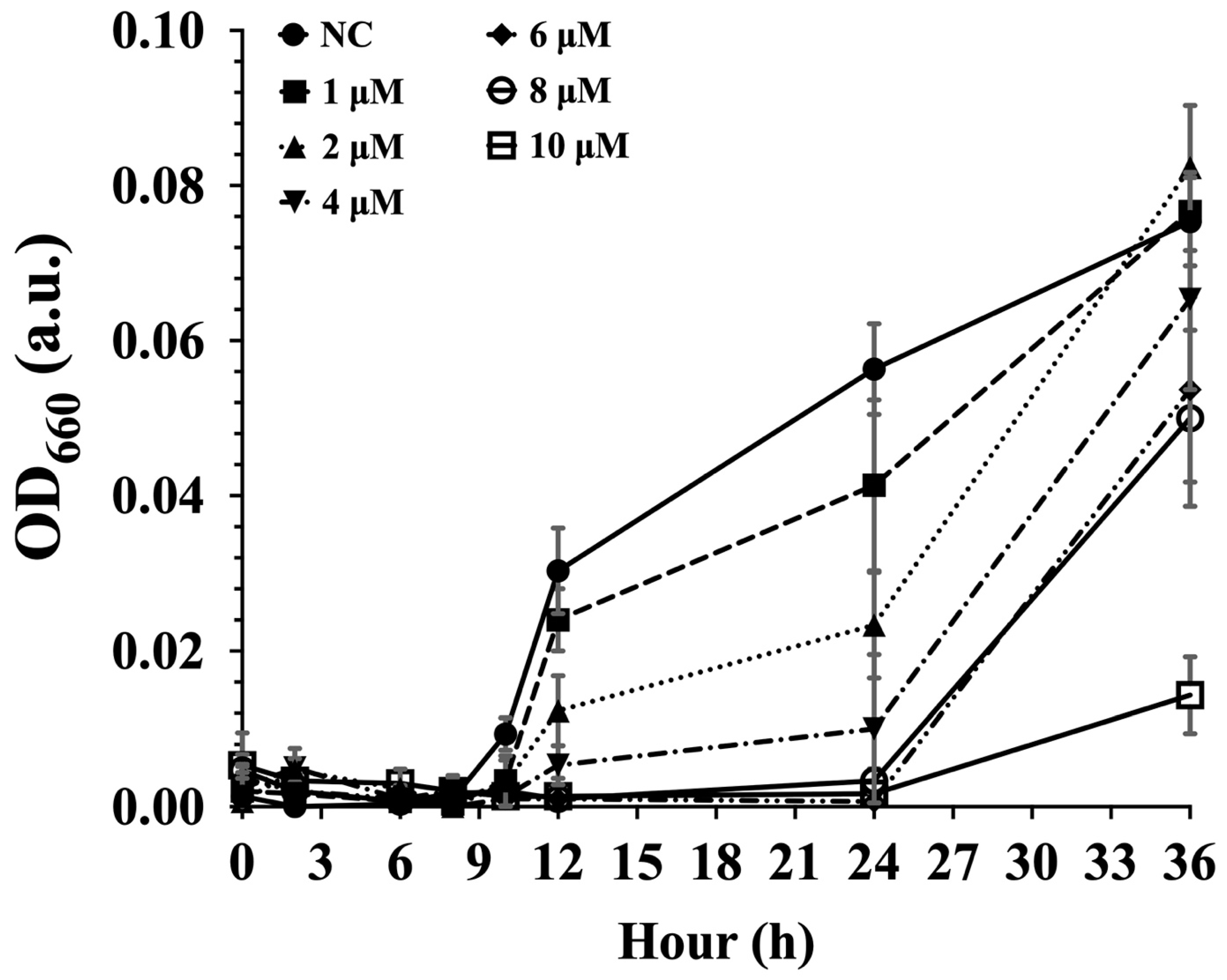
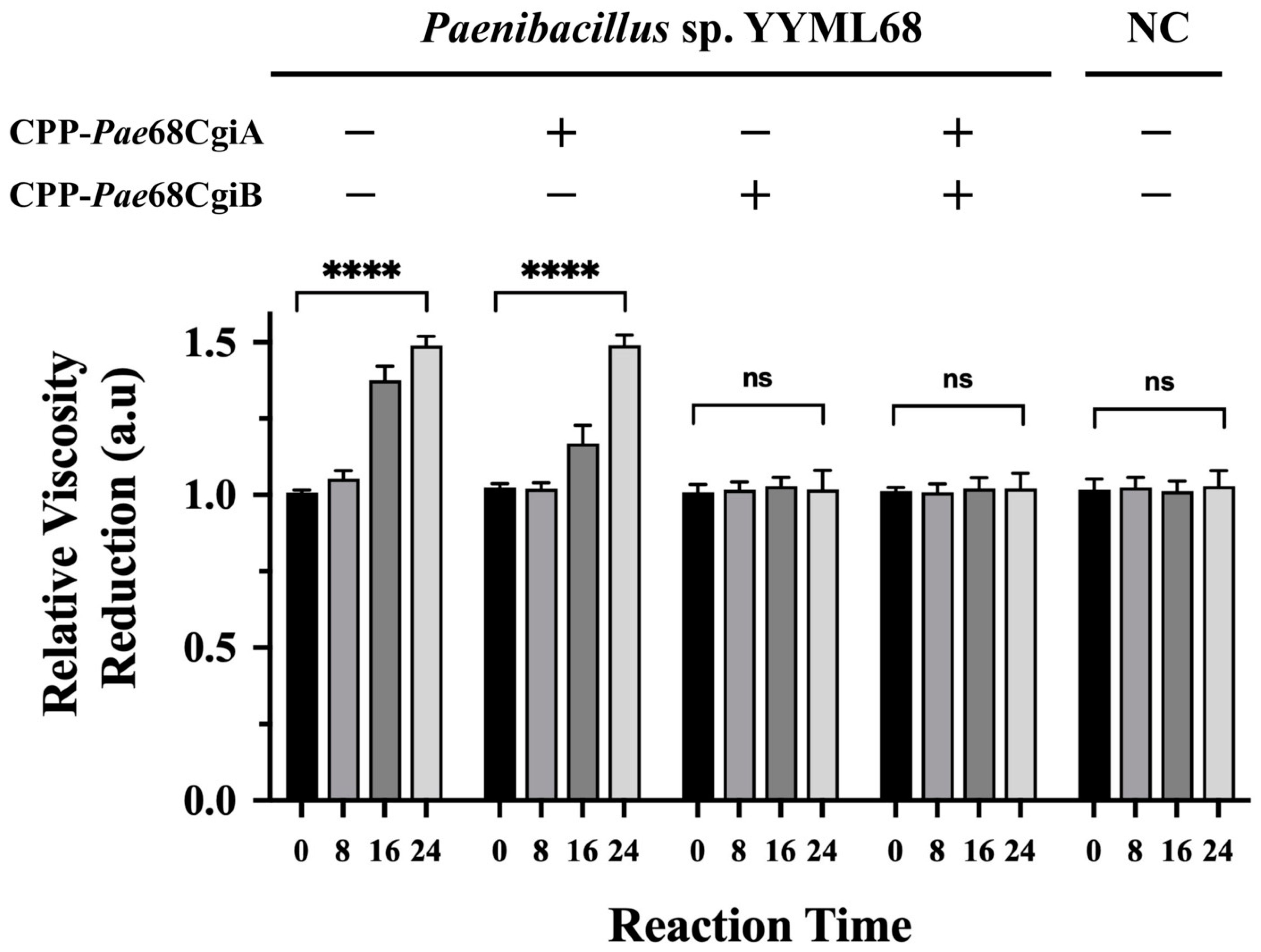
3.2. CPP–PNA Efficiently Suppresses Protein Translation in Strain YYML68
3.3. Carrageenase Gene Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, T.A. Gene Cloning and DNA Analysis: An Introduction, 8th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2021; p. 1. [Google Scholar]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Freudl, R. Signal peptides for recombinant protein secretion in bacterial expression systems. Microb. Cell Factories 2018, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-free gene expression: An expanded repertoire of applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef] [Green Version]
- Souque, C.; Escudero, J.A.; MacLean, R.C. Integron activity accelerates the evolution of antibiotic resistance. Elife 2021, 10, e62474. [Google Scholar] [CrossRef]
- Desler, C.; Durhuus, J.A.; Rasmussen, L.J. Genome-wide screens for expressed hypothetical proteins. Methods Mol. Biol. 2012, 815, 25–38. [Google Scholar]
- Sharan, R.; Ulitsky, I.; Shamir, R. Network-based prediction of protein function. Mol. Syst. Biol. 2007, 3, 88. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Three factors underlying incorrect in silico predictions of essential metabolic genes. BMC Syst Biol 2008, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.E.; Brown, A.L.; Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinform. 2007, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Rembeza, E.; Engqvist, M.K.M. Experimental and computational investigation of enzyme functional annotations uncovers misannotation in the EC 1.1.3.15 enzyme class. PLoS Comput. Biol. 2021, 17, e1009446. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; Liu, H. Enhanced Recombinant Protein Production Under Special Environmental Stress. Front. Microbiol. 2021, 12, 630814. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Maclntyre, L.W.; Brady, S.F. Refactoring biosynthetic gene clusters for heterologous production of microbial natural products. Curr. Opin. Biotechnol. 2021, 69, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Bauman, K.D.; Li, J.; Murata, K.; Mantovani, S.M.; Dahesh, S.; Nizet, V.; Luhavaya, H.; Moore, B.S. Refactoring the Cryptic Streptophenazine Biosynthetic Gene Cluster Unites Phenazine, Polyketide, and Nonribosomal Peptide Biochemistry. Cell Chem. Biol. 2019, 26, 724–736 e727. [Google Scholar] [CrossRef]
- Zhang, X.; Hindra; Elliot, M.A. Unlocking the trove of metabolic treasures: Activating silent biosynthetic gene clusters in bacteria and fungi. Curr. Opin. Microbiol. 2019, 51, 9–15. [Google Scholar] [CrossRef]
- Toyohara, D.; Yokoi, Y.; Inoue, G.; Muraoka, T.; Mori, T. Abiotic Factors Promote Cell Penetrating Peptide Permeability in Enterobacteriaceae Models. Front. Microbiol. 2019, 10, 2534. [Google Scholar] [CrossRef] [Green Version]
- Inoue, G.; Toyohara, D.; Mori, T.; Muraoka, T. Critical Side Chain Effects of Cell-Penetrating Peptides for Transporting Oligo Peptide Nucleic Acids in Bacteria. ACS Appl. Biol. Mater. 2021, 4, 3462–3468. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Vandermies, M.; Joyeux, C.; Martin, R.; Barbeyron, T.; Michel, G.; Vandenbol, M. Discovering novel enzymes by functional screening of plurigenomic libraries from alga-associated Flavobacteriia and Gammaproteobacteria. Microbiol. Res. 2016, 186–187, 52–61. [Google Scholar] [CrossRef]
- Hatada, Y.; Mizuno, M.; Li, Z.; Ohta, Y. Hyper-production and characterization of the iota-carrageenase useful for iota-carrageenan oligosaccharide production from a deep-sea bacterium, Microbulbifer thermotolerans JAMB-A94T, and insight into the unusual catalytic mechanism. Mar. Biotechnol. 2011, 13, 411–422. [Google Scholar] [CrossRef]
- Michel, G.; Chantalat, L.; Fanchon, E.; Henrissat, B.; Kloareg, B.; Dideberg, O. The iota-carrageenase of Alteromonas fortis. A beta-helix fold-containing enzyme for the degradation of a highly polyanionic polysaccharide. J. Biol. Chem. 2001, 276, 40202–40209. [Google Scholar] [CrossRef] [Green Version]
- Michel, G.; Helbert, W.; Kahn, R.; Dideberg, O.; Kloareg, B. The structural bases of the processive degradation of iota-carrageenan, a main cell wall polysaccharide of red algae. J. Mol. Biol. 2003, 334, 421–433. [Google Scholar] [CrossRef]
- Hejtmankova, A.; Vanova, J.; Spanielova, H. Cell-penetrating peptides in the intracellular delivery of viral nanoparticles. Vitam. Horm. 2021, 117, 47–76. [Google Scholar]
- Palm-Apergi, C.; Dowdy, S.F. Protein Delivery by PTDs/CPPs. Methods Mol. Biol. 2022, 2383, 257–264. [Google Scholar] [PubMed]
- Ruter, C. Delivery of Antibiotics by Cell-Penetrating Peptides to Kill Intracellular Pathogens. Methods Mol. Biol. 2022, 2383, 335–345. [Google Scholar] [PubMed]
- Perera, J.D.R.; Carufe, K.E.W.; Glazer, P.M. Peptide nucleic acids and their role in gene regulation and editing. Biopolymers 2021, 112, e23460. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kong, J.; Zhang, X. Application of peptide nucleic acid in electrochemical nucleic acid biosensors. Biopolymers 2021, 112, e23464. [Google Scholar] [CrossRef] [PubMed]
- Nacher-Vazquez, M.; Santos, B.; Azevedo, N.F.; Cerqueira, L. The role of Nucleic Acid Mimics (NAMs) on FISH-based techniques and applications for microbial detection. Microbiol. Res. 2022, 262, 127086. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef]
- Narenji, H.; Teymournejad, O.; Rezaee, M.A.; Taghizadeh, S.; Mehramuz, B.; Aghazadeh, M.; Asgharzadeh, M.; Madhi, M.; Gholizadeh, P.; Ganbarov, K.; et al. Antisense peptide nucleic acids againstftsZ andefaA genes inhibit growth and biofilm formation of Enterococcusfaecalis. Microb. Pathog. 2020, 139, 103907. [Google Scholar] [CrossRef]
- Javanmard, Z.; Kalani, B.S.; Razavi, S.; Farahani, N.N.; Mohammadzadeh, R.; Javanmard, F.; Irajian, G. Evaluation of cell-penetrating peptide-peptide nucleic acid effect in the inhibition of cagA in Helicobacter pylori. Acta Microbiol. Immunol. Hung. 2020, 67, 66–72. [Google Scholar] [CrossRef]
- da Silva, K.E.; Ribeiro, S.M.; Rossato, L.; Dos Santos, C.P.; Preza, S.E.; Cardoso, M.H.; Franco, O.L.; Migliolo, L.; Simionatto, S. Antisense peptide nucleic acid inhibits the growth of KPC-producing Klebsiella pneumoniae strain. Res. Microbiol. 2021, 172, 103837. [Google Scholar] [CrossRef]
- Xue, X.Y.; Mao, X.G.; Zhou, Y.; Chen, Z.; Hu, Y.; Hou, Z.; Li, M.K.; Meng, J.R.; Luo, X.X. Advances in the delivery of antisense oligonucleotides for combating bacterial infectious diseases. Nanomedicine 2018, 14, 745–758. [Google Scholar] [CrossRef]
- Okonkwo, C.C.; Ujor, V.; Cornish, K.; Ezeji, T.C. Inactivation of the Levansucrase Gene in Paenibacillus polymyxa DSM 365 Diminishes Exopolysaccharide Biosynthesis during 2,3-Butanediol Fermentation. Appl. Environ. Microbiol. 2020, 86, e00196-20. [Google Scholar] [CrossRef] [PubMed]
- Descamps, T.; De Smet, L.; De Vos, P.; de Graaf, D.C. Unbiased random mutagenesis contributes to a better understanding of the virulent behaviour of Paenibacillus larvae. J. Appl. Microbiol. 2018, 124, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.D.; Aronstein, K.A. Transformation of the Gram-positive honey bee pathogen, Paenibacillus larvae, by electroporation. J. Microbiol. Methods 2008, 75, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Heinze, S.; Kornberger, P.; Gratz, C.; Schwarz, W.H.; Zverlov, V.V.; Liebl, W. Transmating: Conjugative transfer of a new broad host range expression vector to various Bacillus species using a single protocol. BMC Microbiol. 2018, 18, 56. [Google Scholar] [CrossRef] [Green Version]
- Meliawati, M.; May, T.; Eckerlin, J.; Heinrich, D.; Herold, A.; Schmid, J. Insights in the Complex DegU, DegS, and Spo0A Regulation System of Paenibacillus polymyxa by CRISPR-Cas9-Based Targeted Point Mutations. Appl. Environ. Microbiol. 2022, 88, e0016422. [Google Scholar] [CrossRef]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar] [CrossRef]
- Patel, R.R.; Sundin, G.W.; Yang, C.H.; Wang, J.; Huntley, R.B.; Yuan, X.; Zeng, Q. Exploration of Using Antisense Peptide Nucleic Acid (PNA)-cell Penetrating Peptide (CPP) as a Novel Bactericide against Fire Blight Pathogen Erwinia amylovora. Front. Microbiol. 2017, 8, 687. [Google Scholar] [CrossRef] [Green Version]
- Tailhades, J.; Takizawa, H.; Gait, M.J.; Wellings, D.A.; Wade, J.D.; Aoki, Y.; Shabanpoor, F. Solid-Phase Synthesis of Difficult Purine-Rich PNAs through Selective Hmb Incorporation: Application to the Total Synthesis of Cell Penetrating Peptide-PNAs. Front. Chem. 2017, 5, 81. [Google Scholar] [CrossRef]
- Patil, N.A.; Thombare, V.J.; Li, R.; He, X.; Lu, J.; Yu, H.H.; Wickremasinghe, H.; Pamulapati, K.; Azad, M.A.K.; Velkov, T.; et al. An Efficient Approach for the Design and Synthesis of Antimicrobial Peptide-Peptide Nucleic Acid Conjugates. Front. Chem. 2022, 10, 843163. [Google Scholar] [CrossRef]
- Li, C.; Callahan, A.J.; Phadke, K.S.; Bellaire, B.; Farquhar, C.E.; Zhang, G.; Schissel, C.K.; Mijalis, A.J.; Hartrampf, N.; Loas, A.; et al. Automated Flow Synthesis of Peptide-PNA Conjugates. ACS Cent. Sci. 2022, 8, 205–213. [Google Scholar] [CrossRef]
- Michel, G.; Czjzek, M. Polysaccharide-degrading enzymes from marine bacteria. In Marine Enzymes for Biocatalysis: Sources, Biocatalytic Characteristic and Bioprocesses of Marine Enzymes; Trincone, A., Ed.; Woodhead Publishing Limited: Cambridge, UK, 2013; pp. 429–464. [Google Scholar]
- Antonopoulos, A.; Favetta, P.; Helbert, W.; Lafosse, M. On-line liquid chromatography electrospray ionization mass spectrometry for the characterization of kappa- and iota-carrageenans. Application to the hybrid iota-/nu-carrageenans. Anal. Chem. 2005, 77, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Fatema, M.K.; Nonami, H.; Ducatti, D.R.; Goncalves, A.G.; Duarte, M.E.; Noseda, M.D.; Cerezo, A.S.; Erra-Balsells, R.; Matulewicz, M.C. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry analysis of oligosaccharides and oligosaccharide alditols obtained by hydrolysis of agaroses and carrageenans, two important types of red seaweed polysaccharides. Carbohydr. Res. 2010, 345, 275–283. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoi, Y.; Kawabuchi, Y.; Zulmajdi, A.A.; Tanaka, R.; Shibata, T.; Muraoka, T.; Mori, T. Cell-Penetrating Peptide–Peptide Nucleic Acid Conjugates as a Tool for Protein Functional Elucidation in the Native Bacterium. Molecules 2022, 27, 8944. https://doi.org/10.3390/molecules27248944
Yokoi Y, Kawabuchi Y, Zulmajdi AA, Tanaka R, Shibata T, Muraoka T, Mori T. Cell-Penetrating Peptide–Peptide Nucleic Acid Conjugates as a Tool for Protein Functional Elucidation in the Native Bacterium. Molecules. 2022; 27(24):8944. https://doi.org/10.3390/molecules27248944
Chicago/Turabian StyleYokoi, Yasuhito, Yugo Kawabuchi, Abdullah Adham Zulmajdi, Reiji Tanaka, Toshiyuki Shibata, Takahiro Muraoka, and Tetsushi Mori. 2022. "Cell-Penetrating Peptide–Peptide Nucleic Acid Conjugates as a Tool for Protein Functional Elucidation in the Native Bacterium" Molecules 27, no. 24: 8944. https://doi.org/10.3390/molecules27248944