

In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein
 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. In Vitro Screening
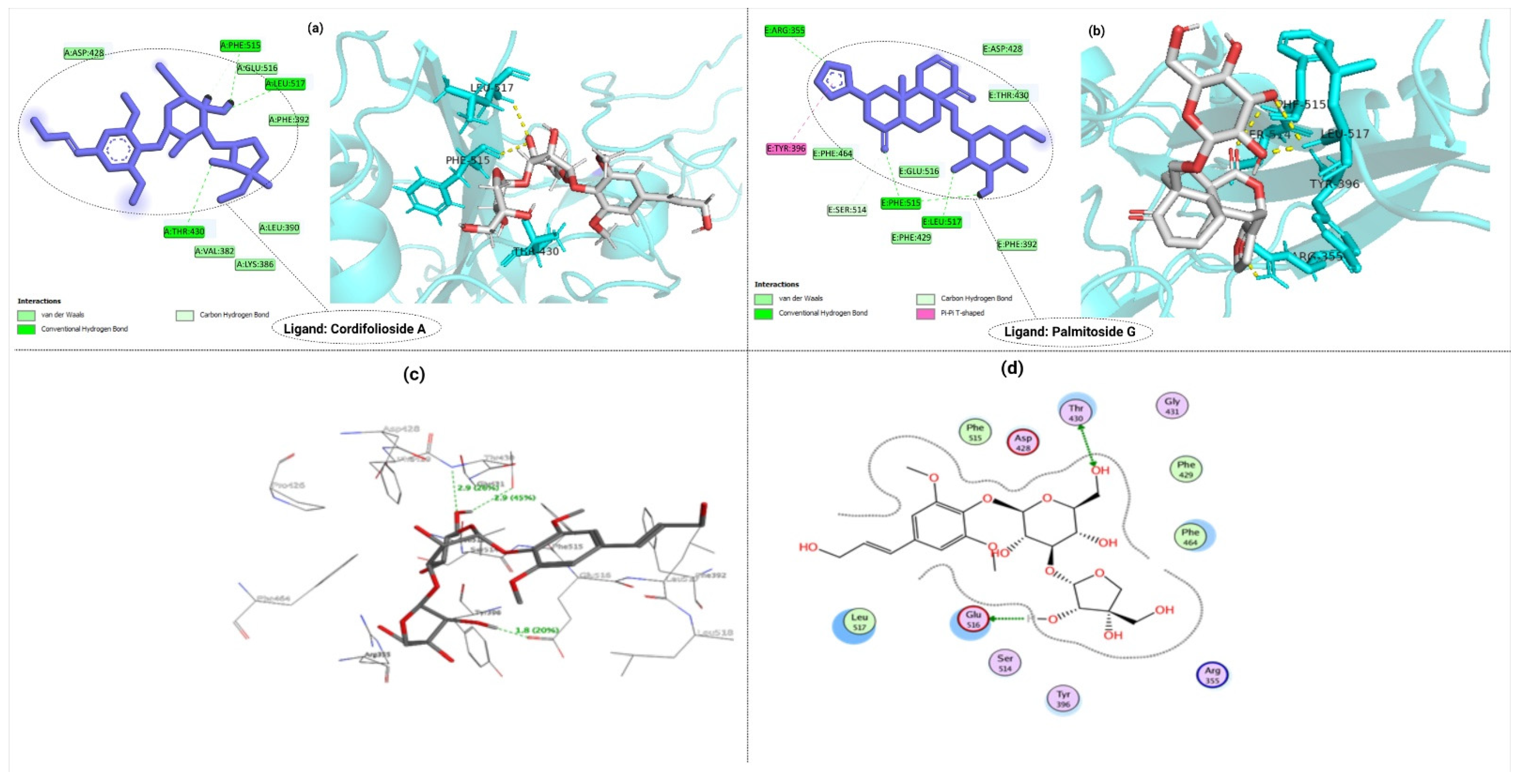
2.2. Molecular Docking of S1-RBD with Ligands
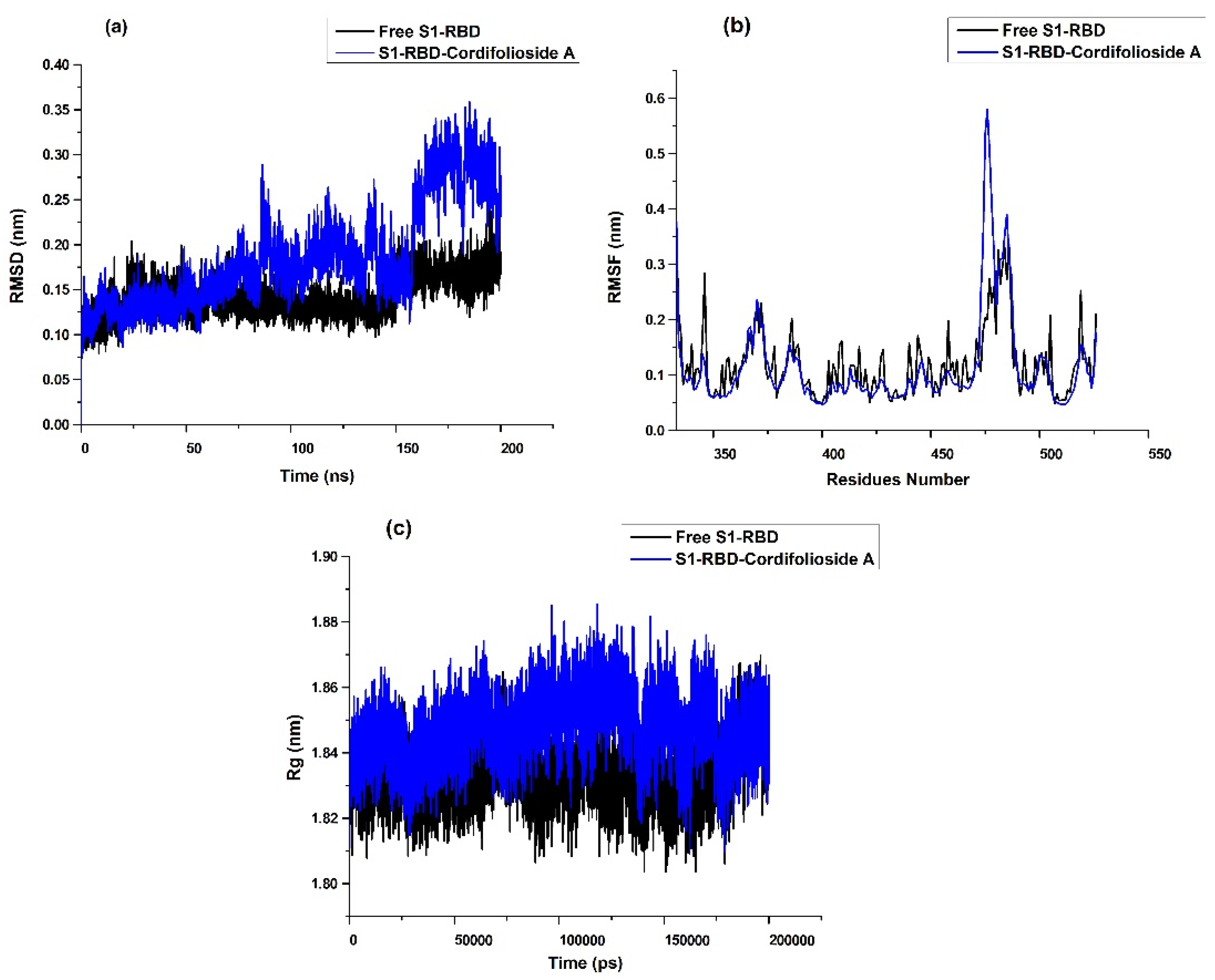
2.3. Molecular Dynamics Simulation Analysis
2.4. Binding Free Energy (BFE) Analysis
2.5. Analysis of ADMET Profiles
3. Discussion
4. Materials and Methods
4.1. In Vitro Spike S1-RBD and hACE2 Inhibitory Activity of SARS-CoV-2 by Enzyme-Linked Immunosorbent Assay (ELISA)
4.2. Computational Workstation
4.3. Protein Preparation
4.4. Preparation of Ligands
4.5. Binding Site Prediction
4.6. Molecular Docking and Validation
4.7. Molecular Dynamics Simulation
4.8. Binding Free Energy Calculation
4.9. Pharmacokinetics Study of Secondary Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adhikari, B.; Marasini, B.P.; Rayamajhee, B.; Bhattarai, B.R.; Lamichhane, G.; Khadayat, K.; Adhikari, A.; Khanal, S.; Parajuli, N. Potential roles of medicinal plants for the treatment of viral diseases focusing on COVID-19: A review. Phytother. Res. 2021, 35, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Afrose, S.; Biswas, S.; Nagata, A.; Paul, G.K.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Zaman, S.; Uddin, M.S.; et al. Plant-derived compounds effectively inhibit the main protease of SARS-CoV-2: An in silico approach. PLoS ONE 2022, 17, e0273341. [Google Scholar] [CrossRef] [PubMed]
- Marahatha, R.; Shrestha, A.; Sharma, K.; Regmi, B.P.; Sharma, K.R.; Poudel, P.; Basnyat, R.C.; Parajuli, N. In silico study of alkaloids: Neferine and berbamine potentially inhibit the SARS-CoV-2 RNA-dependent RNA polymerase. J. Chem. 2022, 2022, e7548802. [Google Scholar] [CrossRef]
- Majumdar, M.; Singh, V.; Misra, T.K.; Roy, D.N. In silico studies on structural inhibition of SARS-CoV-2 main protease Mpro by major secondary metabolites of Andrographis paniculata and Cinchona officinalis. Biologia 2022, 77, 1373–1389. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Alluwaimi, A.M.; Alshubaith, I.H.; Al-Ali, A.M.; Abohelaika, S. The coronaviruses of animals and birds: Their zoonosis, vaccines, and models for SARS-CoV and SARS-CoV-2. Front. Vet. Sci. 2020, 7, 582287. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wunderink, R.G. MERS, SARS and other coronaviruses as causes of pneumonia: MERS, SARS and coronaviruses. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.S. The SARS epidemic in Hong Kong: What lessons have we learned? J. R. Soc. Med. 2003, 96, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. SARS Working Group. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, H.; Gupte, J.; Fletcher, H.; Hammond, L.; Lowe, N.; Pelling, M.; Raina, N.; Shahid, T.; Shanks, K. COVID-19 as a global challenge: Towards an inclusive and sustainable future. Lancet Planet. Health 2020, 4, e312–e314. [Google Scholar] [CrossRef] [PubMed]
- Dilucca, M.; Forcelloni, S.; Georgakilas, A.G.; Giansanti, A.; Pavlopoulou, A. Codon usage and phenotypic divergences of SARS-CoV-2 genes. Viruses 2020, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Puttaswamy, H.; Gowtham, H.G.; Ojha, M.D.; Yadav, A.; Choudhir, G.; Raguraman, V.; Kongkham, B.; Selvaraju, K.; Shareef, S.; Gehlot, P.; et al. In silico studies evidenced the role of structurally diverse plant secondary metabolites in reducing SARS-CoV-2 pathogenesis. Sci. Rep. 2020, 10, 20584. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Ji, M.; Pei, F.; Zhao, Q.; Zhou, Y.; Hong, Y.; Han, S.; Wang, J.; Wang, Q.; et al. Transmission routes analysis of SARS-CoV-2: A systematic review and case report. Front. Cell Dev. Biol. 2020, 8, 618. [Google Scholar] [CrossRef]
- Rolta, R.; Yadav, R.; Salaria, D.; Trivedi, S.; Imran, M.; Sourirajan, A.; Baumler, D.J.; Dev, K. In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: An approach to prevent virus assembly. J. Biomol. Struct. Dyn. 2021, 39, 7017–7034. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Buchholz, U.J.; Bukreyev, A.; Yang, L.; Lamirande, E.W.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. USA 2004, 101, 9804–9809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–23. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, M.; Saha, S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PLoS ONE 2020, 15, e0237300. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, M.-L.; Chien, C.-S.; Yarmishyn, A.A.; Yang, Y.-P.; Lai, W.-Y.; Luo, Y.-H.; Lin, Y.-T.; Chen, Y.-J.; Chang, P.-C.; et al. Highlight of immune pathogenic response and hematopathologic effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 infection. Front. Immunol. 2020, 11, 1022. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Soltane, R.; Abo Elmaaty, A.; Tantawy, M.A.; Antar, S.A.; Yahya, G.; Chrouda, A.; Pashameah, R.A.; Mustafa, M.; Abu Mraheil, M.; et al. Coronavirus disease (COVID-19) control between drug repurposing and vaccination: A comprehensive overview. Vaccines 2021, 9, 1317. [Google Scholar] [CrossRef] [PubMed]
- Ashour, N.A.; Elmaaty, A.A.; Sarhan, A.A.; Elkaeed, E.B.; Moussa, A.M.; Erfan, I.A.; Al-Karmalawy, A.A. A systematic review of the global intervention for SARS-CoV-2 combating: From drugs repurposing to Molnupiravir approval. Drug Des. Dev. Ther. 2022, 16, 685–715. [Google Scholar] [CrossRef]
- CDC. Coronavirus Disease 2019 (COVID-19). Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 11 February 2020).
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature 2021, 596, 273–275. [Google Scholar] [CrossRef]
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef]
- Ganjhu, R.K.; Mudgal, P.P.; Maity, H.; Dowarha, D.; Devadiga, S.; Nag, S.; Arunkumar, G. Herbal plants and plant preparations as remedial approach for viral diseases. VirusDisease 2015, 26, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Alamgeer; Younis, W.; Asif, H.; Sharif, A.; Riaz, H.; Bukhari, I.A.; Assiri, A.M. Traditional medicinal plants used for respiratory disorders in Pakistan: A review of the ethno-medicinal and pharmacological evidence. Chin. Med. 2018, 13, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mintah, S.O.; Asafo-Agyei, T.; Archer, M.-A.; Junior, P.A.-A.; Boamah, D.; Kumadoh, D.; Appiah, A.; Ocloo, A.; Boakye, Y.D.; Agyare, C. Medicinal Plants for Treatment of Prevalent Diseases. In Pharmacognosy-Medicinal Plants; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Nugraha, R.V.; Ridwansyah, H.; Ghozali, M.; Khairani, A.F.; Atik, N. Traditional herbal medicine candidates as complementary treatments for COVID-19: A review of their mechanisms, pros, and cons. J. Evid. Based Complement. Altern. Med. 2020, 2020, 2560645. [Google Scholar] [CrossRef] [PubMed]
- Marahatha, R.; Basnet, S.; Bhattarai, B.R.; Budhathoki, P.; Aryal, B.; Adhikari, B.; Lamichhane, G.; Poudel, D.K.; Parajuli, N. Potential natural inhibitors of xanthine oxidase and HMG-CoA reductase in cholesterol regulation: In silico analysis. BMC Complement. Med. Ther. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Marahatha, R.; Gyawali, K.; Sharma, K.; Gyawali, N.; Tandan, P.; Adhikari, A.; Timilsina, G.; Bhattarai, S.; Lamichhane, G.; Acharya, A.; et al. Pharmacologic activities of phytosteroids in inflammatory diseases: Mechanism of action and therapeutic potentials. Phytother. Res. 2021, 35, 5103–5124. [Google Scholar] [CrossRef]
- Ho, T.T.; Tran, Q.T.; Chai, C.L. The polypharmacology of natural products. Future Med. Chem. 2018, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Drasar, P.B.; Khripach, V.A. Growing importance of natural products research. Molecules 2020, 25, 6. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.E.; Le Sage, V.; Lakdawala, S.S. Viral, and host heterogeneity and their effects on the viral life cycle. Nat. Rev. Microbiol. 2021, 19, 272–282. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Giri, P.; Uniyal, P.L. Edible ferns in India and their medicinal uses: A review. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2022, 92, 17–25. [Google Scholar] [CrossRef]
- Bhatt, A.; Bisht, A.; Rawal, R.; Dhar, U. Assessment of status and biomass of Swertia angustifolia: A high value Himalayan medicinal plant. Afr. J. Plant Sci. 2007, 1, 1–6. [Google Scholar] [CrossRef]
- Khan, M.B.; Rathi, B. Tinospora cordifolia-An immunomodulatory drug in Ayurveda for prevention and treatment of COVID-19. Int. J. Res. Pharm. Sci. 2020, 11, 1695–1699. [Google Scholar] [CrossRef]
- Chhetri, V.T.; Jha, P.; Maharjan, S.K. An ethnomedicinal appraisal of medicinal plants used in COVID-19 pandemic in Buddhabumi municipality, Southern Nepal. Ethnobot. Res. Appl. 2021, 22, 1–19. [Google Scholar]
- Bahadori, M.B.; Dinparast, L.; Zengin, G. The genus Heracleum: A comprehensive review on its phytochemistry, pharmacology, and ethnobotanical values as a useful herb. Compr. Rev. Food Sci. Food Saf. 2016, 15, 1018–1039. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Nath, L.K.; Bhise, S. Antioxidant and antimicrobial activities of Heracleum nepalense D Don root. Trop. J. Pharm. Res. 2005, 4, 341–347. [Google Scholar] [CrossRef]
- Sharov, A.V.; Burkhanova, T.M.; Tok, T.T.; Babashkina, M.G.; Safin, D.A. Computational analysis of Molnupiravir. Int. J. Mol. Sci. 2022, 23, 1508. [Google Scholar] [CrossRef]
- Bouback, T.A.; Pokhrel, S.; Albeshri, A.; Aljohani, A.M.; Samad, A.; Alam, R.; Hossen, M.S.; Al-Ghamdi, K.; Talukder, M.E.K.; Ahammad, F.; et al. Pharmacophore-based virtual screening, quantum mechanics calculations, and molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules 2021, 26, 4961. [Google Scholar] [CrossRef]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 22, bbab098. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Wink, M. Molecular Modes of Action of Cytotoxic Alkaloids: From DNA Intercalation, Spindle Poisoning, Topoisomerase Inhibition to Apoptosis and Multiple Drug Resistance. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 64, pp. 1–47. [Google Scholar] [CrossRef]
- Wink, M. Modes of action of herbal medicines and plant secondary metabolites. Medicines 2015, 2, 251–286. [Google Scholar] [CrossRef] [PubMed]
- Reichling, J. Plant-Microbe Interactions and Secondary Metabolites with Antibacterial, Antifungal and Antiviral Properties. In Annual Plant Reviews Online; American Cancer Society: Atlanta, GA, USA, 2018; pp. 214–347. [Google Scholar] [CrossRef]
- Sharma, U.; Bala, M.; Kumar, N.; Singh, B.; Munshi, R.K.; Bhalerao, S. Immunomodulatory active compounds from Tinospora cordifolia. J. Ethnopharmacol. 2012, 141, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Mishra, N.; Singh, J.; Khanuja, S. Tinospora cordifolia (Guduchi): A reservoir plant for therapeutic applications: A review. Indian J. Tradit. Knowl. 2004, 3, 257–270. [Google Scholar]
- Haque, M.A.; Jantan, I.; Abbas Bukhari, S.N. Tinospora species: An overview of their modulating effects on the immune system. J. Ethnopharmacol. 2017, 207, 67–85. [Google Scholar] [CrossRef]
- Sachan, S.; Dhama, K.; Latheef, S.K.; Abdul Samad, H.; Mariappan, A.K.; Munuswamy, P.; Singh, R.; Singh, K.P.; Malik, Y.S.; Singh, R.K. Immunomodulatory potential of Tinospora cordifolia and CpG ODN (TLR21 Agonist) against the very virulent, infectious Bursal disease virus in SPF chicks. Vaccines 2019, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Khadka, D.; Dhamala, M.K.; Li, F.; Aryal, P.C.; Bhatta, S. The use of medicinal plant to prevent COVID-19 in Nepal. The use of medicinal plants to prevent COVID-19 in Nepal. J. Ethnobiol. Ethnomedicine 2021, 17, 26. [Google Scholar] [CrossRef]
- Khanna, K.; Kohli, S.K.; Kaur, R.; Bhardwaj, A.; Bhardwaj, V.; Ohri, P.; Sharma, A.; Ahmad, A.; Bhardwaj, R.; Ahmad, P. Herbal immune-boosters: Substantial warriors of pandemic COVID-19 battle. Phytomedicine 2021, 85, 153361. [Google Scholar] [CrossRef]
- Tahmasbi, S.F.; Revell, M.A.; Tahmasebi, N. Herbal medication to enhance or modulate viral infections. Nurs. Clin. 2021, 56, 79–89. [Google Scholar] [CrossRef]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Koh, C.C.; Reker, D.; Brown, J.B.; Wang, H.; Lee, N.K.; Liow, H.; Dai, H.; Fan, H.-M.; Chen, L.; et al. Predicting protein-ligand interactions based on bow-pharmacological space and Bayesian additive regression trees. Sci. Rep. 2019, 9, 7703. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Boobbyer, D.N.A.; Goodford, P.J.; McWhinnie, P.M.; Wade, R.C. New hydrogen-bond potentials for use in determining energetically favorable binding sites on molecules of known structure. J. Med. Chem. 1989, 32, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Dolgonosov, A.M. The universal relationship between the energy and length of a covalent bond derived from the theory of generalized charges. Russ. J. Inorg. Chem. 2017, 62, 344–350. [Google Scholar] [CrossRef]
- Srivastava, N.; Garg, P.; Srivastava, P.; Seth, P.K. A molecular dynamics simulation study of the ACE2 receptor with screened natural inhibitors to identify novel drug candidate against COVID-19. PeerJ 2021, 9, e11171. [Google Scholar] [CrossRef]
- Chowdhury, P. In silico investigation of phytoconstituents from Indian medicinal herb, ‘Tinospora cordifolia (giloy)’ against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J. Biomol. Struct. Dyn. 2021, 39, 6792–6809. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Muehlbacher, M.; Spitzer, G.M.; Liedl, K.R.; Kornhuber, J. Qualitative prediction of blood-brain barrier permeability on a large and refined dataset. J. Comput. Aided Mol. Des. 2011, 25, 1095–1106. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Watanabe, R.; Ohashi, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Mizuguchi, K. Development of an in silico prediction system of human renal excretion and clearance from chemical structure information incorporating fraction unbound in plasma as a descriptor. Sci. Rep. 2019, 9, 18782. [Google Scholar] [CrossRef] [Green Version]
- Hillebrecht, A.; Muster, W.; Brigo, A.; Kansy, M.; Weiser, T.; Singer, T. Comparative evaluation of in silico systems for Ames test mutagenicity prediction: Scope and limitations. Chem. Res. Toxicol. 2011, 24, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Mulliner, D.; Schmidt, F.; Stolte, M.; Spirkl, H.-P.; Czich, A.; Amberg, A. Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol. 2016, 29, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, L.; Cheng, F.; Wu, Z.; Bian, H.; Xu, C.; Li, W.; Liu, G.; Shen, X.; Tang, Y. In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model. 2014, 54, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; She, G.; Han, D.; Wang, W.; Liu, Z.; Liu, B. Genus Tinospora: Ethnopharmacology, phytochemistry, and pharmacology. Evid. Based Complement. Altern. Med. 2016, 2016, 9232593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Dwivedee, B.P.; Bisht, D.; Dash, A.K.; Kumar, D. The chemical constituents and diverse pharmacological importance of Tinospora cordifolia. Heliyon 2019, 5, e02437. [Google Scholar] [CrossRef] [Green Version]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A inhibits binding of host ACE2 receptor with spike RBD domain of SARS-CoV-2, a lead compound for blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, A.; Gupta, U.; Sharma, S.; Sharma, A.; Gupta, U. Molecular Docking Studies on the Anti-Fungal Activity of Allium sativum (Garlic) against Mucormycosis (Black Fungus) by BIOVIA Discovery Studio Visualizer 21.1.0.0. Ann. Antivir. Antiretrovir. 2021, 5, 028–032. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.-H.; Chen, P.-H.; Hung, H.-Y.; Hwang, T.-L.; Chiang, C.-C.; Thang, T.D.; Kuo, P.-C.; Wu, T.-S. Chemical constituents from the stems of Tinospora sinensis and their bioactivity. Molecules 2018, 23, 2541. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-J.; Foloppe, N. Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: Comparison to programs MOE and Catalyst. J. Chem. Inf. Model. 2010, 50, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Del Carpio, C.A.; Takahashi, Y.; Sasaki, S. A new approach to the automatic identification of candidates for ligand receptor sites in proteins: (I) Search for pocket regions. J. Mol. Graph. 1993, 11, 23–29. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, R. Classification of current scoring functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Onodera, K.; Satou, K.; Hirota, H. Evaluations of molecular docking programs for virtual screening. J. Chem. Inf. Model. 2007, 47, 1609–1618. [Google Scholar] [CrossRef]
- Okimoto, N.; Futatsugi, N.; Fuji, H.; Suenaga, A.; Morimoto, G.; Yanai, R.; Ohno, Y.; Narumi, T.; Taiji, M. High-performance drug discovery: Computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000528. [Google Scholar] [CrossRef]
- Yue, Y.; Zhao, S.; Sun, Y.; Yan, X.; Liu, J.; Zhang, J. Effects of plant extract aurantio-obtusin on pepsin structure: Spectroscopic characterization and docking simulation. J. Lumin. 2017, 187, 333–339. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Rout, S.; Mahapatra, R.K. In silico screening of novel inhibitors of M17 leucine amino peptidase (LAP) of Plasmodium vivax as therapeutic candidate. Biomed. Pharmacother. 2016, 82, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, S.; Dahal, S.R.; Chaudhary, B.P.; Mohanty, S. Structure and function studies of Asian Corn Borer Ostrinia furnacalis pheromone binding protein2. Sci. Rep. 2018, 8, 17105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mulakala, C.; Viswanadhan, V.N. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J. Mol. Graph. Model. 2013, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.K.; Singh, S.K.; Singh, P.; Chellaperumal, P.; Reddy, K.K.; Selvaraj, C. Exploring the selectivity of a ligand complex with CDK2/CDK1: A molecular dynamics simulation approach: Exploring the selectivity of a ligand complex with CDK2 and CDK1. J. Mol. Recognit. 2012, 25, 504–512. [Google Scholar] [CrossRef]
- González-Medina, M.; Naveja, J.J.; Sánchez-Cruz, N.; Medina-Franco, J.L. Open chemoinformatic resources to explore the structure, properties and chemical space of molecules. RSC Adv. 2017, 7, 54153–54163. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Machhar, J.; Mittal, A.; Agrawal, S.; Pethe, A.M.; Kharkar, P.S. Computational prediction of toxicity of small organic molecules: State-of-the-art. Phys. Sci. Rev. 2019, 4, 20190009. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N. | Plants | Voucher Code | Location (Altitude) | Reported Medicinal Uses | References |
|---|---|---|---|---|---|
| 1 | Dryopteris wallichiana | KHP 03 | Bajhang (2935 m a.s.l) | The rhizome is used as an anti-rheumatic and for treating constipation | [44] |
| 2 | Swertia kingii | KHP 08 | Doti (3071 m a.s.l) | Blood purifier, skin disease, bitter tonic for fever, indigestion, laxative, anthelmintic, antidiarrhoeal, antiperiodic, and bronchial asthma | [45] |
| 3 | Swertia ciliata | KHP 24 | Doti (3127 m a.s.l) | Used as a substitute for Swertia kingii | [45] |
| 4 | Tinospora cordifolia | TUCH 210052 | Bajhang (2907 m a.s.l) | Immunomodulatory, anticancer, antiviral antidiabetic, antimicrobial, antioxidant, anti-inflammatory, antipyretic, and antiallergic | [46,47] |
| 5 | Pogostemon benghalensis | TUCH 210050 | Bajura (3001 m a.s.l) | Antioxidant, anticancer, antibacterial, antifungal, anti-inflammatory, and antiviral | [47] |
| 6 | Justicia adhatoda | TUCH 210051 | Doti (3107 m a.s.l) | Immunomodulatory, antimicrobial, antibacterial, antiviral, anti-inflammatory, and antioxidant | [47] |
| 7 | Heracleum nepalense | TUCH 210059 | Bajura (3143 m a.s.l) | Breath rate stimulator, antidiarrheal, aphrodisiac, blood pressure stimulator, tonic, antioxidant, and antimicrobial | [48,49] |
| Compound | S-Score | GOLD Fitness Score | Binding Free Energy MM/GBSA (ΔGbind) (kcal/mol) | Interacting Residues | Interaction Length (Å) |
|---|---|---|---|---|---|
| Molnupiravir | −2.9291 | - | - | Arg346 Glu340 Val341 Asn354 Ser399 Lys356 | 2.84/2.99/4.36 2.05 4.95 4.16 3.07/2.40/1.96 4.73 |
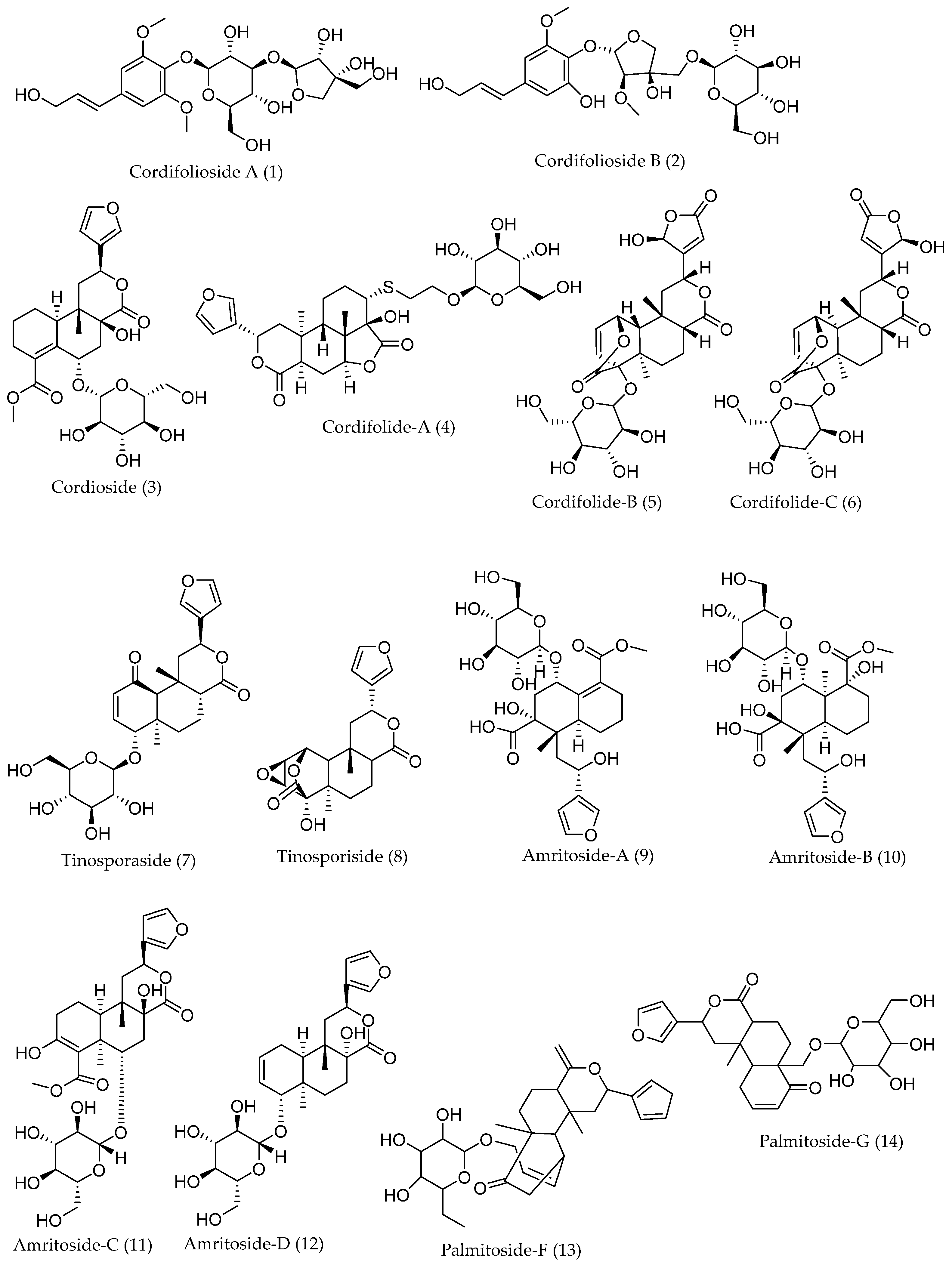
| Cordifolioside A (1) | −7.9942 | 58.27 | −25.09 | Thr430 Phe515 Leu517 | 2.36 2.40/2.83 2.65 |
| Palmitoside G (14) | −7.1871 | 50.80 | −21.23 | Arg355 Tyr396 Ser514 Phe515 Leu517 | 2.37 4.92 3.26 2.83/2.90 1.91 |
| S1-RBD-Complex | No. of Hydrogen Bonds | Interacting Residues | Bond Length (Å) | Hydrogen Bond Strength |
|---|---|---|---|---|
| S-Cordifolioside-A complex | 1 | Phe515 | 2.4 | 20% |
| S1-RBD Binding Site | Size of Amino Acids | Residues |
|---|---|---|
| 1 | 36 | Arg454, Phe456, Arg457, Lys458, Ser459, Asp467, Ser469, Thr470, Glu471, Ile472, Tyr473, Gln474, Cyc480, Asn481, Gly482, Pro491 |
| 2 | 59 | Arg355, Tyr380, Gly381, Val382, Leu390, Phe392, Tyr396, Pro426, Asp428, Phe429, Thr430, Gly431, Phe464, Leu513, Ser514, Phe515, Glu516 |
| 3 | 27 | Arg403, Glu406, Lys417, Tyr453, Ser494, Tyr453, Ser494, Try495, Gly496, Phe497, Gln498, Asn501, Tyr505 |
| 4 | 18 | Cyc336, Pro337, Phe338, Gly339, Phe342, Val367, Leu368, Ser371, Phe374 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basnet, S.; Marahatha, R.; Shrestha, A.; Bhattarai, S.; Katuwal, S.; Sharma, K.R.; Marasini, B.P.; Dahal, S.R.; Basnyat, R.C.; Patching, S.G.; et al. In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein. Molecules 2022, 27, 8957. https://doi.org/10.3390/molecules27248957
Basnet S, Marahatha R, Shrestha A, Bhattarai S, Katuwal S, Sharma KR, Marasini BP, Dahal SR, Basnyat RC, Patching SG, et al. In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein. Molecules. 2022; 27(24):8957. https://doi.org/10.3390/molecules27248957
Chicago/Turabian StyleBasnet, Saroj, Rishab Marahatha, Asmita Shrestha, Salyan Bhattarai, Saurav Katuwal, Khaga Raj Sharma, Bishnu P. Marasini, Salik Ram Dahal, Ram Chandra Basnyat, Simon G. Patching, and et al. 2022. "In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein" Molecules 27, no. 24: 8957. https://doi.org/10.3390/molecules27248957
APA StyleBasnet, S., Marahatha, R., Shrestha, A., Bhattarai, S., Katuwal, S., Sharma, K. R., Marasini, B. P., Dahal, S. R., Basnyat, R. C., Patching, S. G., & Parajuli, N. (2022). In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein. Molecules, 27(24), 8957. https://doi.org/10.3390/molecules27248957