Abstract
The construction of an N–C chiral axis for N-aryl indole derivatives is meaningful as they widely exist in functionalized molecules. This work provides a novel method for this purpose via amination of amino acid derivatives at the C2 position of the indole and chiral center induced chiral axis formation. The protocol of this transformation is easily accessible, not requiring metal or an organic chiral catalyst, endowing this method with great potential in the construction of axis chiral N-aryl indoles.
1. Introduction
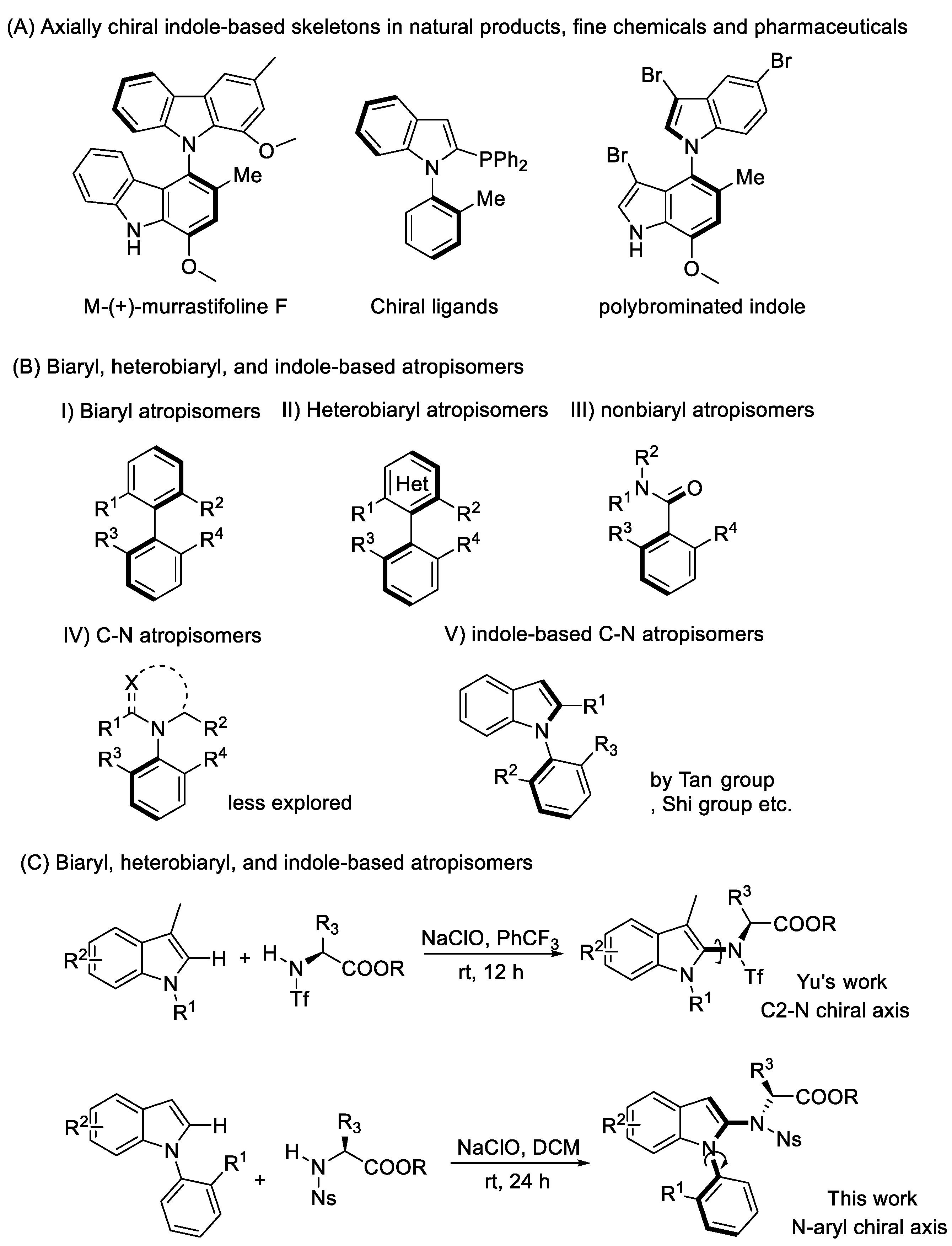
Atropisomers around the C–N chiral axis, which have a typical chiral axis connecting a substituted nitrogen and the aryl-related hindrance, are one of the most important classes of axially chiral compounds. For example, Murrastifoline F was isolated from murraya alkaloid, which possesses anti-HIV activity [1,2,3]. Moreover, such skeletons have been used as chiral organocatalysts and ligands in enantioselective reactions, due to their specific electronic properties among biaryls (Scheme 1a) [4,5,6].
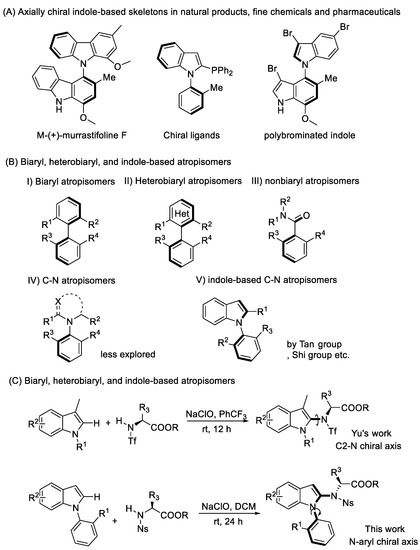
Scheme 1.
Functionalized axially chiral indole-based molecules and outline for indole axially chiral construction.
Given the importance of C−N axially chiral biaryls in pharmaceuticals, organic synthesis, and materials, catalytic asymmetric syntheses of these C–N axially chiral compounds and their applications to asymmetric transformations are of current interest [7,8,9,10,11,12,13,14,15,16,17]. Additionally, it would be meaningful to introduce axial chirality into the indole skeleton, as the indole skeleton is widely apparent in many natural products and drug molecules. Catalytic asymmetric synthesis of indole-based heterobiaryl axially chiral compounds was disclosed by the Shi [18,19,20], Tan [21], Fu [22,23], and Yan [24] groups, etc. The chiral axis was normally built at the C2 or C3 position of the indole ring [25,26,27,28,29,30,31], while recent literature has reported the stereoselective synthesis of axially chiral indole derivatives possessing a sterically hindered phenyl group on the nitrogen atom [32,33,34,35]. Despite these promising achievements, asymmetric synthesis of indole-based atropisomers, especially with an N–C chiral axis, is still a synthetic challenge and remains largely unexplored (Scheme 1b).
This study tackles this synthetic defiance considering the asymmetric catalysis free and easy formation of indole-based heterobiaryl axially chiral compounds, in line with strategies such as chiral center induced chiral axis formation and the central-to-axial chirality conversion [36,37,38]. It is worth noting that the Yu’s group described a series of examples for atroposelective coupling of indoles at the C2 position with chiral amino acid-based sulfonamides via a central-to-axial chirality conversion strategy [39,40]. Encouraged by that, we rationalized that the construction of an N–C chiral axis for N-aryl indole derivatives possessing a sterically hindered N-phenyl group via amination at the C2 position of the indole and sequence chiral center induced chiral axis formation would represent a novel strategy for constructing indole-based N–C atropisomers (Scheme 1c).
2. Results and Discussion
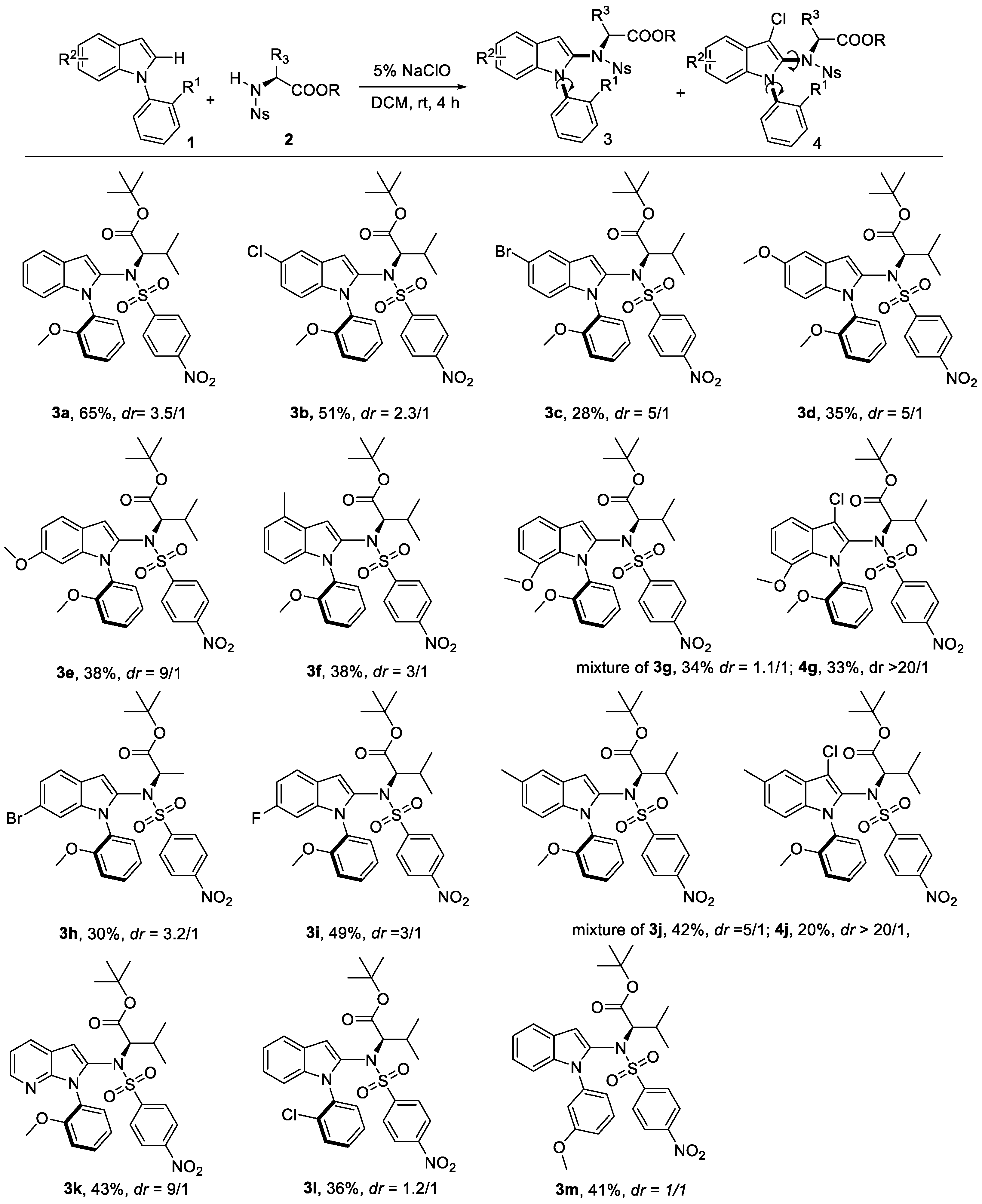
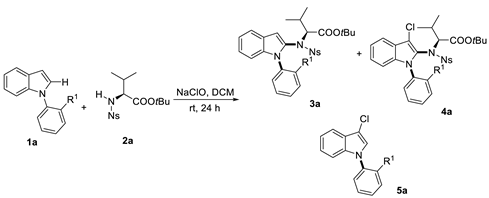
To start this work, N-phenyl indole 1a and tert-butyl ((4-nitrophenyl)sulfonyl)-L-valinate 2a were used as the model substrate to test the feasibility for the C–N amination (Table 1). Product 3a was afforded using an aqueous NaClO solution with 1a in different solvents. The solvent was also crucial to both the yield and diastereoisomers (dr) value. The optimal solvent was found to be DCM in 65% yield and 3.5/1 dr value (entry 1–6). Only a trace product was observed in the protic solvent (entry 4). The yield loss was due to the formation of byproduct 4a and oxidative byproduct 5a. Interestingly, biaxial chirality was observed as the chlorine atom brings steric hindrance at the C3 position. In Yu’s work, excellent dr value was observed at the C–N axis chirality at the indole C2 position. Similar high dr was tracked for compound 4a (entry 1).

Table 1.
Solvent screening for indole C–N axially chiral construction.
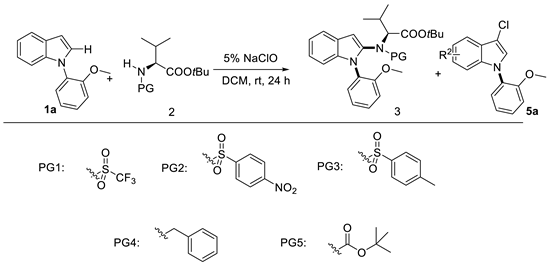
In should be mentioned that the protecting group of valinate also significantly affected the yield and dr (Table 2). The p-nitrobenzene sulfonyl group (Ns) was favorable for this transformation (entry 2). Other sulfonyl protecting groups yielded the product 3a in less than 5% yield. The benzyl protecting group (Bn) and tert-butoxycarbonyl group (Boc) were conductive to produce byproduct 5a.
Table 2.
Protecting group screening for indole C–N axially chiral construction.
In order to show the generality of this strategy, the couplings of various indoles with N-Ns protected amino acid derivatives were investigated under the optimal conditions. First, a series of substituted indole derivatives at different positions reacted with 2a to give product 3a–3j in moderate yield (Scheme 2, 3a–3h). The electron-donating group bearing indoles at the C4-C5 position had lower yields but improved dr value (3d, 3e vs. 3a, 3b, 3i). For substrate 3j, the C-3 chlorination product 4j was produced at the same time. The dr value of 3j was around 5/1, but its side product 4j had excellent dr value. A similar dr value with the model substrate 3a was obtained for C-6 substituted indoles (3h, 3i). One example using the heterocyclic indole analogue with 1-(2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine was also conducted with 43% yield and good dr value (3k). Next, we explored the variations at the N-aryl ring of the indole. A less bulky ortho-substituted substrate resulted in lower dr value (3l). In the case of substitutions at the meta position, 1:1 of two diastereomers were observed (3m).
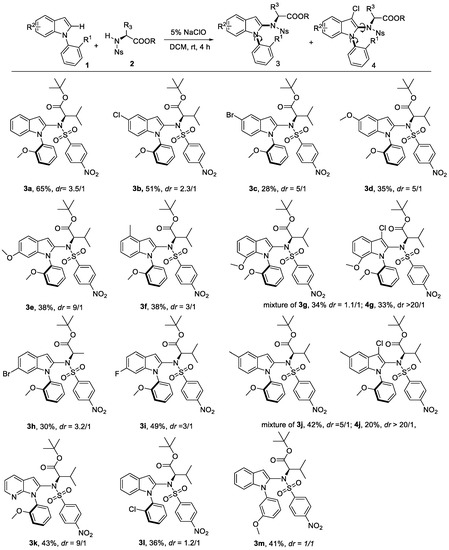
Scheme 2.
Substrates scope for various 1-(2-methoxyphenyl)-1H-indoles for construction of C–N axially chirality. reaction conditions: indoles (0.2 mmol), amino acid (0.6 mmol), 5% NaClO (0.6 mmol), DCM (4 mL), rt, 24 h.
After that, we continued to investigate the reactivity and stereoselectivity of the reactions of N-phenylindole (1a) with various amino acid derivatives (Scheme 3). Generally, the steric hindrance of amino acid derivatives had a positive influence on the stereoselectivity. To our delight, excellent dr value was shown with amino acid derivatives such as L-Isoleucine (3p), L-Norvaline (3q). But the yield was slightly decreased with bulker amino acid. As for the L-Cyclohexyl glycine derivative 3s and 2-Amino-1-propanol derivative 3t, byproducts 4s and 4t were tracked, respectively.
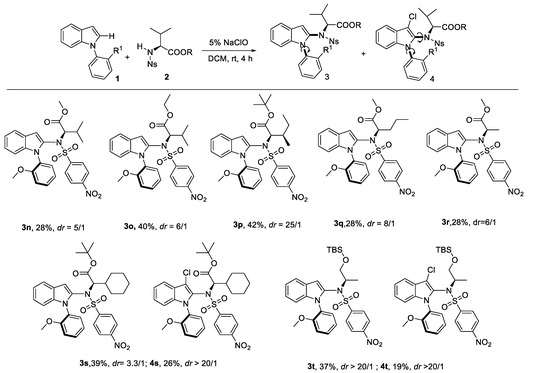
Scheme 3.
Substrates scope for various amino acid for construction of C–N axially chirality. reaction conditions: indoles (0.2 mmol), amino acid (0.6 mmol), 5% NaClO (0.6 mmol), DCM (4 mL), rt, 24 h.
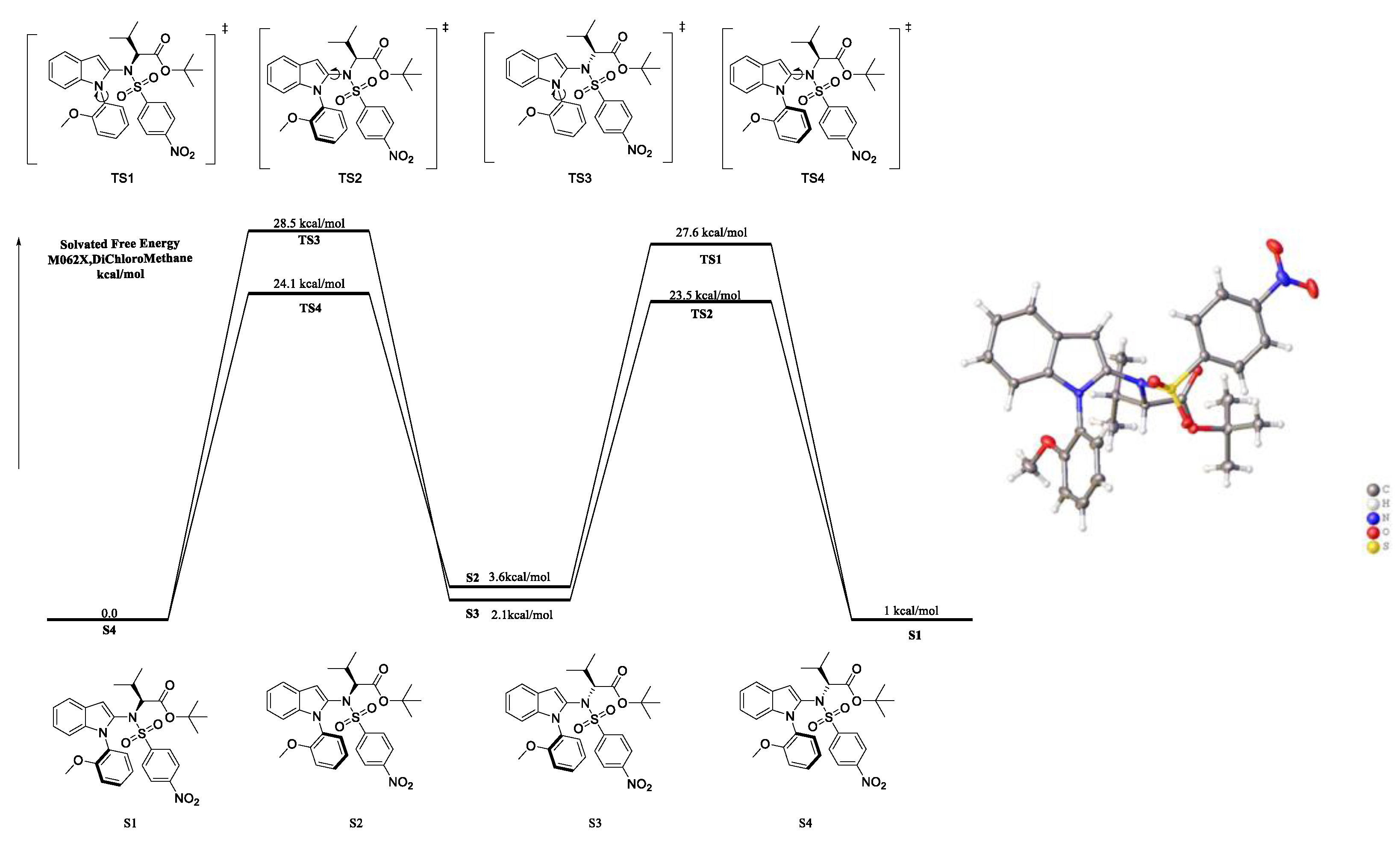
The absolute configuration of axially chiral product 3a was determined to be (Ra,S) by single-crystal X-ray diffraction analysis (Scheme 4) (CCDC 2222365 for the major diastereomer of 3a, see the Supporting Information for details). Thus, the absolute configurations of other axially chiral products were assigned to be (Ra,S) by analogy with 3a. Next, we investigated the configurational stability and the rotational barrier of product 3a and other isomers (S1–S4, Scheme 4) via DFT calculations (Scheme 4). It was discovered that the indole C2–N axis was unstable (the rotate barrier was 24.1 kcal/mol and 23.5 kcal/mol, respectively, for TS2 and TS4), which will rapidly disappear under room temperature. However, the N–Caryl axis had good configurational stability due to the fact that the rotational barrier was high (28.5 kcal/mol for TS3, 27.6 kcal/mol for TS1). As the amino acid was optically pure, the diastereo ratio may come from the atropisomerism of an N–Caryl axis.
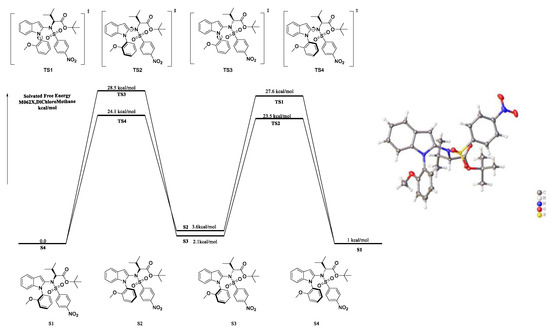
Scheme 4.
DFT calculations for rotate barrier for C–N axis and N-aryl axis and absolute configuration of 3a.
In summary, we present here an atroposelective coupling of indoles with chiral amino acid-based sulfonamides mediated by hypo-halides through chiral center induced chiral axis formation strategy. The reaction delivers 2-amido-N-arylindoles with an N–C chiral axis in a moderate to good dr value. The substrates and reaction protocol of this transformation are easily accessible, not requiring chiral catalyst, endowing this method with great potential in the construction of axis chiral N-arylindoles.
3. Materials and Methods
Unless otherwise noted, all reactants or reagents including dry solvents were obtained from commercial suppliers and used as received. NaClO (Sodium hypochlorite solution reagent grade, available chlorine 4.00–4.99%) was purchased from Lingfeng reagent company, Shanghai, China. All the reactions were conducted using reaction tube (10 mL) under argon atmosphere. Analytical thin layer chromatography (TLC) was performed using Silica Gel 60 F25 plates. Column chromatograph was performed on silica gel 100~200 mesh. 1H and 13C NMR spectra were obtained in CDCl3 or DMSO using 300 MHz, 400 MHz Varian NMR spectrometer. Chemical shifts in 1H NMR spectra are reported in parts per million (ppm) on the δ scale from an internal standard of residual CDCl3 (7.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constant in Hertz (Hz). Chemical shifts in 13C NMR spectra are reported in ppm on the δ scale from the central peak of residual CDCl3 (77.16 ppm).
3.1. General Procedures for Indole C2 Amination with N-Ns Protected Amino Acid
A 10 mL round bottom flask was equipped with a rubber septum and magnetic stir bar and was charged with 1 (0.2 mmol), 2 (0.6 mmol), NaClO (0.6 mmol, 774 μL, 5% in water) in DCM (4 mL) at room temperature for 24 h. Upon completion of the reaction, the mixture was washed with saturated Na2CO3 aqueous solution (4 mL), water, and saturated brine (4 mL) in sequence. The organic layer was concentrated and purified via a flash column (PE/EA from 50/1 to 30/1).
3.2. Characterization Data for Compounds
tert-butyl N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate 3a. According to the general procedure, purification by flash chromatography, 3a (75.4 mg, 65%, dr = 3.5:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, CDCl3) δ 8.33 (d, J = 9.0 Hz, 2H × 0.78), 8.26 (d, J = 9.0 Hz, 2H × 0.22), 7.99 (d, J = 9.0 Hz, 2H × 0.78), 7.83 (d, J = 9.0 Hz, 2H × 0.22),7.56–7.37 (m, 3H), 7.14–6.89 (m, 4H), 6.57–6.54 (m, 1H), 6.01 (s, 1H × 0.22), 6.00 ((s, 1H × 0.78), 3.91 (d, J = 9.0 Hz, 1H × 0.78)), 3.56 (d, J = 9.0 Hz, 1H × 0.22), 3.50 (s, 3H × 0.78), 3.47 (s, 1H × 0.22), 1.55–1.41 (m, 1H × 0.78), 1.05 (s, 9H × 0.22), 0.94 (s, 9H × 0.78), 0.54 (d, J = 6.6 Hz, 3H × 0.22), 0.47 (d, J = 6.5 Hz, 3H × 0.78), 0.14 (d, J = 6.7 Hz, 3H × 0.78). 13C NMR (75 MHz, Chloroform-d) δ 167.4, 156.5, 150.5, 145.0, 137.2, 132.7, 130.6, 125.0, 124.8, 123.8, 123.5, 121.3, 121.0, 120.5, 111.7, 111.4, 104.6, 82.3, 72.1, 55.3, 29.1, 27.9, 19.6, 19.5. HRMS Calcd. For C30H33N3NaO7S [M+Na]+: 602.1931; found: 602.1920.
tert-butyl N-(5-chloro-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate 3b. According to the general procedure, purification by flash chromatography, 3b (66.2 mg, 51%, dr = 2.3:1) was obtained as a yellow solid from 1b (0.2 mmol, 51.5 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.51 (d, J = 6.0 Hz, 2H × 0.70), 8.42 (d, J = 6.0 Hz, 2H × 0.30), 8.23 (d, J = 6.0 Hz, 2H × 0.7), 8.16–8.12 (m, 1H), 8.04 (d, J = 6.0 Hz, 2H × 0.3), 7.64–7.59 (m, 2H), 7.46–7.38 (m, 1H), 7.32–7.16 (m, 3H), 6.88 (d, J = 8.7 Hz, 1H), 6.19 (s, 1H × 0.32), 6.10 (1H × 0.76), 4.33–4.17 (m, 1H), 3.82–3.79 (m, 3H), 1.82–1.79 (m, 1H × 0.70), 1.43 (s, 9H × 0.30), 1.31 (s, 9H × 0.70), 1.13–1.09 (m, 3H × 0.30), 0.90 (d, J = 9.0 Hz, 3H × 0.30), 0.81 (d, J = 9.0 Hz, 3H × 0.3), 0.55 (d, J = 9.0 Hz, 3H × 0.70). 13C NMR (75 MHz, Chloroform-d) δ 167.3, 156.2, 150.4, 144.8, 135.2, 132.4, 132.1, 131.0, 130.8, 130.5, 128.7, 126.2, 126.1, 125.7, 124.3, 124.2, 123.9, 123.8, 123.4, 121.4, 120.3, 120.2, 113.0, 111.5, 104.1, 82.4, 77.4, 71.8, 66.0, 55.5, 55.3, 30.4, 29.2, 28.1, 27.9, 27.9, 27.8, 19.7, 19.4, 19.2. HRMS Calcd. For C30H32ClN3NaO7S [M+Na]+: 636.1542; found: 636.1530.
tert-butyl N-(5-bromo-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate 3c According to the general procedure, purification by flash chromatography, 3c (3.88 mg, 28%, dr =5:1) was obtained as a yellow solid from 1c (0.2 mmol, 60.4 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.39 (d, J = 9.0 Hz, 2H × 0.83), 8.34 (d, J = 9.0 Hz, 2H × 0.17), 8.30 (d, J = 9.0 Hz, 2H × 0.17) 8.12–8.00 (m, 2H × 0.83), 8.02 (d, J = 9.0 Hz, 2H × 0.17), 7.91 (d, J = 6.0 Hz, 2H), 7.86 (d, J = 3.0 Hz, 1H × 0.83), 7.66 (d, J = 3.0 Hz, 1H × 0.17), 7.53–7.47 (m, 1H), 7.42 (dd, J = 9.0 Hz, 3.0 Hz, 1H × 0.83), 7.32 (dd, J = 9.0 Hz, 3.0 Hz, 1H × 0.17), 7.19–7.14 (m, 1H), 7.07–7.04 (m, 1H), 6.73 (d, J = 8.7 Hz, 1H × 0.17), 6.62 (d, J = 8.7 Hz, 1H × 0.83), 6.05 (s, 1H × 0.17), 5.98 (s, 1H × 0.83), 4.20 (d, J = 9.2 Hz, 1H × 0.83), 4.07 (d, J = 9.2 Hz, 1H × 0.17), 3.70 (s, 3H × 0.17), 3.67 (s, 3H × 0.83), 2.15–1.58 (m, 1H), 1.31 (s, 9H × 0.17), 1.20 (s, 9H × 0.83), 1.00–0.42 (m, 6H). Crude 13C NMR (101 MHz, Chloroform-d) δ 171.2, 168.9, 167.3, 156.2, 150.4, 150.2, 144.8, 136.0, 132.4, 131.9, 131.9, 131.7, 131.0, 130.9, 130.5, 129.6, 129.6, 128.7, 127.3, 126.5, 124.2, 124.2, 123.8, 123.4, 123.3, 121.4, 113.8, 113.4, 111.5, 104.0, 103.7, 84.1, 84.0, 82.4, 82.3, 71.8, 66.0, 60.5, 55.5, 55.3, 53.6, 30.4, 29.2, 28.1, 28.0, 27.9, 27.9, 19.7, 19.4, 19.4, 19.2, 14.3. HRMS of 3c Calcd. For C30H32BrN3NaO7S [M+Na]+: 680.1037; found: 680.1032.
tert-butyl N-(5-methoxy-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3d. According to the general procedure, purification by flash chromatography, 3d (42.7 mg, 35%, dr =5:1) was obtained as a yellow solid from 1d (0.2 mmol, 50.7 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.39 (d, J = 9.0 Hz, 2H × 0.83), 8.30 (d, J = 9.0 Hz, 2H × 0.17), 8.14 (d, J = 9.0 Hz, 2H × 0.83), 7.92 (d, J = 9.0 Hz, 2H × 0.17), 7.56–7.45 (m, 2H), 7.36 (t, J = 2.2 Hz, 1H), 7.18–7.11 (m, 1H), 7.04 (dd, J = 8.3, 1.3 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.86–6.83 (m, 1H), 6.74 (d, J = 8.9 Hz, 1H), 6.03 (s, 1H × 0.17), 5.92 (s, 1H × 0.8), 4.21 (d, J = 9.4 Hz, 1H), 3.84 (s, 3H), 3.68 (s, 3H), 1.74–1.71 (m, 1H), 1.29 (s, 9H), 0.97 (d, J = 6.0 Hz, 3H × 0.17), 0.79 (d, J = 6.0 Hz, 3H × 0.17), 0.70 (d, J = 6.0 Hz, 3H × 0.83), 0.43 (d, J = 6.0 Hz, 3H × 0.83). 13C NMR (101 MHz, Chloroform-d) δ 167.4, 157.5, 156.4, 150.4, 145.1, 137.8, 132.7, 130.6, 130.6, 129.8, 124.9, 123.7, 121.7, 121.4, 119.1, 111.5, 110.8, 104.7, 94.7, 82.2, 72.1, 55.7, 55.3, 29.1, 28.0, 27.9, 19.6, 19.5. HRMS Calcd. For C31H35N3NaO8S [M+Na]+: 632.2037; found: 632.2037.
tert-butyl N-(6-methoxy-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3e. According to the general procedure, purification by flash chromatography, 3e (46.3 mg, 38%, dr =9:1) was obtained as a yellow solid from 1e (0.2 mmol, 50.7 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.40 (d, J = 9.0 Hz, 2H × 0.90), 8.31(d, J = 9.0 Hz, 2H × 0.1) 7.95 (d, J = 6.0 Hz, 1H), 7.59–7.49 (m, 1H), 7.40 (d, J = 8.6 Hz, 1H × 0.9), 7.34 (d, J = 8.6 Hz, 1H × 0.1), 7.20 (d, J = 1.3 Hz, 1H), 7.07 (dd, J = 8.4, 1.3 Hz, 1H), 6.81 (dd, J = 8.7, 2.3 Hz, 1H), 6.29 (d, J = 2.3 Hz, 1H), 6.03 (s, 1H × 0.1), 5.93 (d, J = 0.72 Hz, 1H × 0.90), 4.22 (d, J = 9.4 Hz, 1H × 0.9), 3.97 (d, J = 9.4 Hz, 1H × 0.1), 3.74 (s, 3H), 3.71 (s, 3H), 1.77–1.70 (m, 1H), 1.33–1.21 (m, 9H), 0.84–0.43 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 167.45, 157.55, 156.41, 150.35, 145.08, 137.84, 132.70, 130.60, 130.56, 129.82, 128.77, 124.85, 124.31, 123.73, 121.78, 121.71, 121.38, 119.12, 111.47, 110.84, 104.72, 94.68, 82.18, 77.48, 77.16, 76.84, 72.09, 55.74, 55.30, 29.07, 27.99, 27.87, 19.64, 19.49, 19.21. HRMS Calcd. For C31H35N3NaO8S [M+Na]+: 632.2037; found: 632.2037.
tert-butyl N-(1-(2-methoxyphenyl)-4-methyl-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3f. According to the general procedure, purification by flash chromatography, 3f (45.1 mg, 38%, dr =3:1) was obtained as a yellow solid from 1f (0.2 mmol, 47.5 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.57–8.48 (m, 2H), 8.32–8.05 (m, 3H), 7.65 (d, J = 7.8 Hz, 1H), 7.36–7.21 (m, 3H), 7.10–7.08 (m, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.30 (s, 1H × 0.25), 6.17 (s, 1H × 0.75), 4.34 (d, J = 9.6 Hz, 1H), 3.85 (s, 3H), 2.57 (s, 3H), 1.97–1.87 (m, 1H), 1.49 (s, 9H × 0.25), 1.38 (s, 9H × 0.75), 1.17 (d, J = 6.0 Hz, 3H × 0.25), 0.97 (d, J = 6.0 Hz, 3H × 0.25), 0.90 (d, J = 6.0 Hz, 3H × 0.75), 0.61 (d, J = 6.0 Hz, 3H × 0.75). 13C NMR (75 MHz, Chloroform-d) δ 167.4, 156.5, 150.4, 145.0, 136.8, 132.6, 130.8, 130.7, 130.6, 130.4, 130.3, 125.0, 124.9, 123.6, 121.3, 120.7, 120.6, 111.4, 109.4, 103.1, 82.2, 72.2, 58.6, 55.3, 29.0, 28.0, 27.9, 19.6, 19.5, 18.7, 18.6. HRMS Calcd. For C31H35N3NaO7S [M+Na]+: 616.2088; found: 616.2074.
tert-butyl N-(7-methoxy-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate 3g. According to the general procedure, purification by flash chromatography, 3g (37.4 mg, 34 %, dr = 1.1:1) was obtained as a yellow solid from 1g (0.2 mmol, 50.6 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). Crude 1H NMR of 3g. 1H NMR (300 MHz, Chloroform-d) δ 8.40 (d, J = 8.8 Hz, 2H × 0.52), 8.30 (d, J = 8.5 Hz, 2H × 0.48), 8.14–7.94 (m, 2H), 7.43–7.34 (m, 2H), 7.19–6.89 (m, 5H), 6.66 (d, J = 7.8 Hz, 1H), 6.15 (s, 1H × 0.48), 6.01 (s, 1H × 0.52), 4.26–4.07 (m, 1H), 3.79 (s, 3H × 0.48), 3.79 (s, 3H × 0.52), 3.50 (s, 3H × 0.52), 3.50 (s, 3H × 0.48), 1.75–1.60 (m, 1H), 1.35 (s, 9H × 0.48), 1.23 (s, 9H × 0.52), 0.95–0.41 (m, 6H). Crude 13C NMR (75 MHz, Chloroform-d) δ 167.4, 157.3, 155.8, 150.2, 150.0, 147.6, 145.7, 144.9, 131.4, 131.0, 130.6, 130.3, 130.1, 129.7, 129.3, 127.5, 127.1, 126.8, 124.7, 124.6, 123.6, 123.1, 120.6, 120.5, 120.3, 120.1, 117.2, 115.9, 113.9, 111.4, 110.1, 105.2, 105.1, 104.9, 82.1, 71.9, 56.4, 55.9, 55.1, 53.5, 29.0, 27.9, 27.8, 19.6, 19.4, 1.1. HRMS of 3g Calcd. For C31H35N3NaO8S [M+Na]+: 632.2037; found: 632.2024.
tert-butyl N-(6-bromo-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-alaninate 3h. According to the general procedure, purification by flash chromatography, 3h (37.8 mg, 30%, dr =3.2:1) was obtained as a yellow solid from 1h (0.2 mmol, 60.4 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.38 (d, J = 9.0 Hz, 2H × 0.76), 8.30 (d, J = 9.0 Hz, 2H × 0.24), 8.10 (d, J = 9.0 Hz, 2H × 0.76), 8.03–8.78 (m, 1H × 1.48), 7.58–7.46 (m, 1H), 7.40–7.36 (m, 1H), 7.28–7.21 (m, 1H), 7.21–6.99 (m, 3H), 6.09 (s, 1H × 0.23), 6.00 (s, 1H), 4.21–4.05 (m, 1H), 3.72 (s, 3H × 0.23), 3.69 (s, 3H × 0.77), 1.30 (s, 9H × 0.23), 1.19 (s, 9H × 0.74), 1.01–0.41(m, 6H). 13C NMR (101 MHz, Chloroform-d) δ 167.4, 156.3, 150.4, 144.8, 138.0, 132.5, 131.5, 131.0, 130.6, 129.4, 128.8, 124.2, 123.8, 122.6, 121.4, 120.6, 114.6, 111.6, 104.8, 87.9, 82.4, 71.9, 55.4, 29.2, 28.0, 27.9, 19.7, 19.4. HRMS of 3h Calcd. For C30H32BrN3NaO7S [M+Na]+: 680.1037; found: 680.1026.
tert-butyl N-(6-fluoro-1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3i. According to the general procedure, purification by flash chromatography, 3i (58.6 mg, 49%, dr =2.9:1) was obtained as a yellow solid from 1i (0.2 mmol, 48.3 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). HPLC retention time and peak area %: 4.54 min of 71.39%, and 5.05 min of 24.63%. 1H NMR (300 MHz, Chloroform-d) δ 8.39 (d, J = 9.3 Hz, 2H × 0.26), 8.34 (d, J = 9.3 Hz, 2H × 0.26), 8.12 (d, J = 8.9 Hz, 2H × 0.74), 8.02 (d, J = 8.9 Hz, 2H × 0.74), 7.92 (d, J = 7.6 Hz, 1H), 7.53–7.40 (m, 2H), 7.17 (m, 1H), 7.06 (d, J = 9.7 Hz, 1H), 6.89 (m, 1H), 6.52 (dd, J = 9.8, 2.3 Hz, 1H), 6.10 (s, 1H × 0.26), 6.00 (s, 1H × 0.74), 4.20 (d, J = 9.4 Hz, 1H × 0.74), 4.07 (d, J = 9.4 Hz, 1H × 0.76), 3.69 (s, 3H × 0.74), 2.93 (s, 1H × 0.26), 2.15–2.05 (m, 1H × 0.26), 1.74–1.66 (m, 1H × 0.74), 1.26 (s, 9H × 0.24), 1.19 (s, 9H × 0.76), 0.99 (d, J = 3.0 Hz, 1H), 0.78 (d, J = 6.6 Hz, 3H × 0.24), 0.69 (d, J = 6.6 Hz, 3H × 0.76), 0.42 (d, J = 6.6 Hz, 3H × 0.74). 13C NMR (75 MHz, Chloroform-d) δ 168.9, 167.4, 162.4, 159.3, 156.2, 150.4, 150.0, 144.9, 137.2, 137.0, 132.4, 131.3, 131.0, 130.8, 130.5, 128.7, 124.4, 124.3, 123.8, 122.1, 121.9, 121.4, 121.3, 111.5, 109.7, 109.4, 104.7, 98.1, 97.8, 82.3, 77.4, 71.9, 66.0, 55.3, 30.4, 29.1, 27.9, 27.8, 19.6, 19.2. HRMS Calcd. For C30H32FN3NaO7S [M+Na]+: 620.1837; found: 620.1826.
tert-butyl N-(1-(2-methoxyphenyl)-5-methyl-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3j. According to the general procedure, purification by flash chromatography, 3j (49.9 mg, 42%, dr =5:1) and 4j (23.7 mg, 20%, dr > 20:1) was obtained as a yellow solid from 1j (0.2 mmol, 47.5 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). HPLC retention time and peak area %: 3.48 min of 2.05%, 3.82 min of 25.50%, 4.18 min of 4.11% and 4.83 min of 62.76%. Crude 1H NMR of 3j and 4j (300 MHz, Chloroform-d) δ 8.38–8.26 (m, 2.5H), 8.13–7.89 (m, 2.5H), 7.50–7.43 (m, 2H), 7.35–7.28 (m, 2H), 7.18–6.99 (m, 5H), 6.74 (d, J = 8.4 Hz, 1H), 6.42 (d, J = 8.4 Hz, 0.5 H), 6.01 (s, 1H × 0.16), 5.90 (s, 1H × 0.83), 4.21–4.05 (m, 1.3H), 3.79–3.67 (m, 4H), 2.42 (s, 3H), 2.38 (s, 1H), 1.78–1.66 (m, 1H), 1.32–1.20 (m, 12H), 1.01–0.42 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 167.42, 156.46, 155.54, 150.32, 145.05, 139.11, 135.36, 134.16, 132.63, 132.23, 130.99, 130.80, 130.63, 130.47, 129.70, 129.44, 128.75, 125.36, 125.21, 125.00, 124.95, 124.27, 123.72, 121.31, 121.28, 120.45, 112.73, 111.40, 110.44, 104.18, 82.20, 72.01, 56.01, 55.30, 29.08, 27.99, 27.95, 27.88, 21.54, 21.16, 19.67, 19.47. HRMS Calcd. For C31H35N3NaO7S [M+Na]+: 616.2088; found: 616.2075.
tert-butyl N-(1-(2-methoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3k. According to the general procedure, purification by flash chromatography, 3k (49.9 mg, 43 %, dr = 9:1) was obtained as a yellow solid from 1k (0.2 mmol, 44.9 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.40–8.38 (m, 3H × 0.9), 8.35–8.28 (m, 3H × 0.1), 8.10–8.05 (d, J = 7.9 Hz, 2H × 0.9), 8.03–7,85 (m, 2H × 0.1),7.53–7.47 (m, 1H), 7. 20–7.05 (m, 3H), 6.20 (s, 1H × 0.1), 6.09 (s, 1H × 0.9), 4.21 (d, J = 9.2 Hz, 1H), 3.66 (s, 3H × 0.1), 3.66 (s, 3H × 0.9), 1.79–1.69 (m, 1H), 1.32 (s, 9H × 0.1), 1.22 (s, 9H × 0.9), 0.79–0.44 (m, 6H). 13C NMR (75 MHz, Chloroform-d) δ 167.6, 156.6, 150.4, 147.5, 145.6, 144.8, 132.3, 132.1, 131.1, 130.5, 129.5, 123.9, 123.9, 121.3, 118.1, 117.1, 112.0, 103.4, 82.4, 71.6, 55.7, 29.2, 28.0, 27.9, 19.8, 19.4. HRMS Calcd. For C29H33N4O7S [M+H]+:581.2064; found: 581.2050.
tert-butyl N-(1-(2-chlorophenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3l. According to the general procedure, purification by flash chromatography, 3l (42.0 mg, 36%, dr = 1.2:1) was obtained as a yellow solid from 1l (0.2 mmol, 45.5 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.43–8.40 (m, 2H × 0.55), 8.40–8.30 (m, 2H × 0.45), 8.17–8.11 (m, 3H), 7.62–7.48 (m, 4H), 7.22–7.12 (m, 1H), 6.90–6.84 (m, 1H), 6.09 (s, 1H × 0.45), 5.91 (s, 1H × 0.55), 4.23 (m, 1H), 0.89–0.79 (m, 1H), 1.31 (s, 9H × 0.45), 1.15 (s, 9H × 0.55), 0.83–0.75 (m, 3H), 0.83–0.52 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 167.2, 150.5, 150.2, 144.6, 136.1, 134.0, 133.4, 130.8, 130.8, 130.5, 130.5, 128.3, 125.2, 124.9, 124.1, 124.1, 123.8, 123.2, 121.3, 121.2, 121.1, 121.0, 111.8, 105.2, 82.6, 73.1, 53.6, 29.8, 29.1, 28.0, 27.8, 19.8, 18.7. HRMS Calcd. For C29H30ClN3O6S [M+H]+:606.1436; found: 606.1434.
tert-butyl N-(1-(3-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3m. According to the general procedure, purification by flash chromatography, 3m (47.5 mg, 41%) was obtained as a yellow solid from 1m (0.2 mmol, 44.7 mg), 2a (0.6 mmol, 215.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.39 (d, J = 8.6 Hz, 2H), 8.12 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.26–7.10 (m, 5H), 7.07–7.02 (m, 1H), 6.06 (s, 1H), 4.29 (d, J = 8.5 Hz, 1H), 3.87 (s, 3H), 1.65–1.63 (m, 1H), 1.19 (s, 9H), 0.59 (dd, J = 20.8, 6.6 Hz, 6H). 13C NMR (75 MHz, Chloroform-d) δ 167.4, 160.5, 150.4, 145.1, 137.5, 136.8, 131.3, 130.5, 129.9, 125.2, 123.9, 121.5, 121.1, 120.9, 114.7, 111.5, 104.9, 82.5, 71.5, 55.8, 30.3, 27.9, 20.1, 19.0.HRMS Calcd. For C30H33N3NaO7S [M+Na]+: 602.19314; found: 602.1919.
methyl N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3n. According to the general procedure, purification by flash chromatography, 3n (30.1 mg, 28%, dr = 5:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2n (0.6 mmol, 189.8 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.439 (d, J = 9.0 Hz, 2H × 0.83), 8.33 (d, J = 9.0 Hz, 2H × 0.17), 8.09 (d, J = 9.0 Hz, 2H × 0.83), 8.03 (d, J = 9.0 Hz, 2H × 0.17), 7.82 (d, J = 7.8 Hz, 1H), 7.55–7.47 (m, 2H), 7.21–7.02 (m, 4H), 6.83 (d, J = 8.4 Hz, 1H), 6.02 (s, 1H × 0.16), 5.96 (s, 1H × 0.83), 4.31 (d, J = 9.9 Hz, 1H), 3.70 (1H × 0.16), 3.67 (s, 9H × 0.83), 3.46 (s, 3H × 0.16), 3.36 (s, 3H × 0.83), 1.86–1.78 (m, 1H), 0.88–0.45 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 168.7, 156.6, 150.5, 144.3, 132.5, 130.6, 125.1, 124.7, 121.3, 121.0, 120.2, 111.7, 111.4, 72.1, 55.6, 51.7, 28.6, 19.6. HRMS Calcd. For C31H35N3NaO7S [M+Na]+: 616.2088; found: 616.2074.
ethyl N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-valinate3o. According to the general procedure, purification by flash chromatography, 3o (44.1 mg, 40%, dr = 6:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2o (0.6 mmol, 198.2 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.40–8.37 (m, 2H × 0.86), 8.40–8.37 (m, 2H × 0.14), 8.13–7.86 (m, 3H), 7.53–7.47 (m, 2H), 7.21–7.03 (m, 4H), 6.84 (d, J = 9.6 Hz, 1H), 6.03 (s, 1H × 0.14), 5.94 (s, 1H × 0.86), 4.30 (d, J = 10.0 Hz, 1H), 3.92–3.79 (m, 1H), 3.71 (s, 3H × 0.14), 3.67 (s, 3H × 0.86), 3.29 (t, J = 7.1 Hz, 1H), 1.87–1.71 (m, 1H), 1.16–0.89 (m, 3H), 0.80–0.43 (m, 6H). Crude 13C NMR (75 MHz, Chloroform-d) δ 168.7, 156.9, 150.9, 145.0, 137.4, 132.9, 131.6, 131.1, 128.6, 125.4, 125.1, 124.9, 124.1, 124.0, 121.7, 121.4, 120.9, 112.1, 111.9, 103.8, 61.4, 55.7, 42.7, 30.2, 29.1, 20.1, 19.9, 14.7, 14.2. HRMS Calcd. For C28H29N3NaO7S [M+Na]+: 574.1618; found: 574.1602.
tert-butyl N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-isoleucinate 3p. According to the general procedure, purification by flash chromatography, 3p (49.9 mg, 42%, dr = 25:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2p (0.6 mmol, 223.5 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.37 (d, J = 8.7 Hz, 2H), 8.07 (d, J = 8.7 Hz, 2H), 8.00 (d, J = 7.9 Hz, 1H), 7.56–7.47 (m, 2H), 7.21–7.07 (m, 4H), 6.84 (d, J = 8.3 Hz, 1H), 6.27 (s, 1H), 4.40 (d, J = 6.6 Hz, 1H), 3.67 (s, 3H), 1.50–1.42 (m, 1H), 1.23 (s, 9H), 1.09–0.88 (m, 2H), 0.57–0.48 (m, 6H).13C NMR (101 MHz, Chloroform-d) δ 167.9, 156.0, 150.3, 145.0, 136.9, 132.2, 131.1, 130.6, 130.3, 125.0, 124.8, 123.7, 123.4, 121.3, 121.0, 120.4, 111.9, 111.5, 105.2, 82.2, 69.4, 55.2, 35.7, 28.0, 27.2, 15.7, 11.5.HRMS Calcd. For C31H35N3NaO7S [M+Na]+: 616.2088; found: 616.2076.
methyl (R)-2-((N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-4-nitrophenyl)sulfonamido)pentanoate3q. According to the general procedure, purification by flash chromatography, 3q (30.1 mg, 28%, dr = 8:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2q (0.6 mmol, 189.8 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.41 (d, J = 8.8 Hz, 2H), 8.09 (d, J = 8.8 Hz, 2H), 7.78–7,75 (m, 1H), 7.7–7.51 (m, 2H), 7.27–7.16 (m, 3H), 7.09 (d, J = 7.5 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.38 (s, 1H × 0.89), 6.27 (s, 1H × 0.11), 4.61 (t, J = 7.4 Hz, 1H), 3.71 (s, 3H × 0.89), 3.70 (s, 1H × 0.11), 3.52 (s, 3H × 0.11), 3.43 (s, 3H × 0.89), 1.88–0.72 (m, 6H). 13C NMR (75 MHz, Chloroform-d) δ 170.8, 170.0, 156.0, 150.3, 144.6, 136.9, 132.0, 131.3, 130.5, 130.3, 125.2, 124.6, 123.8, 123.6, 123.6, 121.2, 121.0, 120.7, 120.6, 111.7, 111.5, 111.5, 103.8, 64.4, 55.4, 55.2, 52.4, 52.0, 32.0, 20.1, 19.3, 14.3, 13.9. HRMS Calcd. For C27H27N3NaO7S [M+Na]+: 560.1462; found: 560.1452.
methyl N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-N-((4-nitrophenyl)sulfonyl)-D-alaninate3r. According to the general procedure, purification by flash chromatography, 3r (28.5 mg, 28%, dr = 6:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2r (0.6 mmol, 173.0 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.34–8.24 (m, 2H), 7.99 (d, J = 9.0 Hz, 2H × 0.86), 7.91 (d, J = 9.0 Hz, 2H × 0.13), 7.66–7.46 (m, 3H), 7.20–7.02 (m, 4H), 6.86 (d, J = 8.1 Hz, 1H), 6.51 (s, 1H × 0.86), 6.33 (1H × 0.14), 4.73 (q, J = 7.3 Hz, 1H), 3.63 (s, 3H), 3.58 (s, 3H × 0.14), 3.48 (s, 3H × 0.86), 1.19 (d, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 170.7, 156.0, 150.3, 144.8, 137.0, 131.8, 130.4, 125.3, 124.6, 123.7, 121.2, 120.6, 111.8, 111.4, 104.4, 55.3, 52.4, 15.7. HRMS Calcd. For C25H23N3NaO7S [M+Na]+: 532.1149; found: 532.1137.
tert-butyl (R)-2-cyclohexyl-2-((N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-4-nitrophenyl)sulfonamido)acetate3s and tert-butyl (R)-2-((N-(3-chloro-1-(2-methoxyphenyl)-1H-indol-2-yl)-4-nitrophenyl)sulfonamido)-2-cyclohexylacetate 4s. According to the general procedure, purification by flash chromatography, 3s (48.3 mg, 39%, dr = 5:1) and 4s (34.0 mg, 26%, >20:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2s (0.6 mmol, 239.1 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). 1H NMR (300 MHz, Chloroform-d) δ 8.32–8.25 (m, 2H × 0.83), 8.25–8.21 (m, 2H × 0.17), 8.08–8.05 (m, 2H × 0.83), 8.08–8.05 (m, 2H × 0.17),8.05–7.93 (m, 1H × 0.55, from 4s), 7.84–7.82 (m, 1H), 7.62–7.59 (m, 1H), 7.43–7.17 (m, 5H), 7.14–6.93 (m, 8H), 6.76–6.73 (m, 1.5 H), 6.46–6.43 (m, 1 H × 0.55 from 4s), 5.96 (s, 1H × 0.17), 5.71 (s, 1H × 0.83), 4.20 (m, 1H), 3.71 (s, 3H × 0.55 from 4s), 3.65 (s, 3H × 0.83), 3.65 (s, 3H × 0.17), 1.63–13.2 (m, 9H), 1.10(s, 9H × 0.55, from 4s), 1.09 (s, 9H × 0.17), 1.07 (s, 9H × 0.83), 0.90–0.42 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 168.4, 167.0, 156.0, 155.5, 150.3, 145.0, 141.4, 136.7, 132.3, 131.8, 131.1, 131.0, 130.6, 130.3, 129.4, 129.1, 124.8, 124.8, 124.6, 124.2, 123.8, 123.5, 123.4, 121.4, 121.3, 121.2, 120.9, 120.8, 120.5, 120.4, 112.7, 111.7, 111.7, 110.6, 104.2, 82.2, 72.3, 56.0, 55.5, 55.2, 37.7, 30.2, 29.9, 27.9, 27.7, 26.3, 26.2, 26.1. HRMS Calcd. For C33H37N3NaO7S [M+Na]+: 642.2244; found: 642.2231.
(R)-N-(1-((tert-butyldimethylsilyl)oxy)propan-2-yl)-N-(1-(2-methoxyphenyl)-1H-indol-2-yl)-4-nitrobenzenesulfonamide 3t and (R)-N-(1-((tert-butyldimethylsilyl)oxy)propan-2-yl)-N-(3-chloro-1-(2-methoxyphenyl)-1H-indol-2-yl)-4-nitrobenzenesulfonamide 4t. According to the general procedure, purification by flash chromatography, 3t (44.1 mg, 37%, dr > 20:1) and 4t (23.3 mg, 19%, dr > 20:1) was obtained as a yellow solid from 1a (0.2 mmol, 44.6 mg), 2v (0.6 mmol, 224.7 mg), and NaClO (0.6 mmol, 774 μL, 5% in water). Crude 13H NMR of 3t and 4t (300 MHz, Chloroform-d) δ 8.44–8.34 (m, 2H, from 3t), 8.44–8.34 (m, 1H, from 4t), 8.17–8.09 (m, 3H, from 3t and 4t), 7.91–7.84 (m, 1H from 3t, 0.5H from 4t), 7.62–7.47 (m, 2H from 3t, 1H from 4t), 7.20–7.05 (m, 4H from 3t, 2H from 4t), 6.8 (m, 1H from 3t, 0.5H from 4t), 6.03 (s, 1H, from 3t), 4.08–3.87 (m, 1H from 3t, 0.5H from 4t), 3.70 (s, 3H from 3t), 3.67 (s, 1.5H from 4t), 3.43–2.5 (m, 2H from 3t, 1H from 4t), 0.96–0.86 (m, 3H from 3t, 1.5H from 4t), 0.78 (s, 9H from 3t, 4,5H from 4t), −0.11 (s, 6H from 3t), −0.14 (s, 3H from 4t). Crude 13C NMR of 3t and 4t (75 MHz, Chloroform-d) δ 156.3, 155.8, 150.4, 146.7, 145.5, 136.5, 135.6, 132.2, 131.9, 130.9, 130.3, 130.2, 130.0, 129.6, 127.7, 125.1, 124.8, 124.5, 124.5, 124.3, 124.1, 123.5, 123.4, 121.5, 121.2, 121.1, 120.9, 120.6, 118.8, 112.0, 111.8, 111.7, 111.4, 103.2, 77.4, 65.9, 65.4, 61.4, 58.9, 55.5, 55.4, 25.9, 25.8, 18.2, 16.4, 15.7, −5.3, −5.4, −5.4. HRMS of 3t Calcd. For C30H37N3NaO6SSi [M+Na]+: 618.2065; found: 618.2068. HRMS of 4t Calcd. For C30H36ClN3NaO6SSi [M+Na]+: 652.1675; found: 652.1687.
Supplementary Materials
1H and 13 C NMR of all compounds.can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27249008/s1.
Author Contributions
Conceptualization, Y.W. and J.F.; methodology, Y.W., J.Y. and Y.J.; software, Z.W.; formal analysis, Y.W. and J.F.; investigation, Y.W.; resources, Z.T. and C.D.; data curation, Y.W.; writing—review and editing, Y.W. and J.F.; supervision, J.F., T.L. and Y.C.; project administration, J.F.; funding acquisition, J.F. and Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Program Nos. 81973182 and 81673301) and “Double First-Class” University Project from China Pharmaceutical University (Program No. CPU2018GF02).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Hui-Min Xu of The Public Laboratory Platform at China Pharmaceutical University for assistance with NMR techniques.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Xu, D.; Huang, S.; Hu, F.; Peng, L.; Jia, S.; Mao, H.; Gong, X.; Li, F.; Qin, W.; Yan, H. Diversity-Oriented Enantioselective Construction of Atropisomeric Heterobiaryls and N-Aryl Indoles via Vinylidene Ortho-Quinone Methides. CCS Chem. 2022, 4, 2686–2697. [Google Scholar] [CrossRef]
- Bringmann, G.; Tasler, S.; Endress, H.; Kraus, J.; Messer, K.; Wohlfarth, M.; Lobin, W. Murrastifoline-F: First Total Synthesis, Atropo-Enantiomer Resolution, and Stereoanalysis of an Axially Chiral N,C-Coupled Biaryl Alkaloid. J. Am. Chem. Soc. 2001, 123, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Tajuddeen, N.; Bringmann, G. N,C-Coupled naphthylisoquinoline alkaloids: A versatile new class of axially chiral natural products. Nat. Prod. Rep. 2021, 38, 2154–2186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.K.; Xiang, S.H.; Li, S.; Ye, L.; Tan, B. Recent Advances in Catalytic Asymmetric Construction of Atropisomers. Chem. Rev. 2021, 121, 4805–4902. [Google Scholar] [CrossRef]
- Rodriguez-Salamanca, P.; Fernandez, R.; Hornillos, V.; Lassaletta, J.M. Asymmetric Synthesis of Axially Chiral C-N Atropisomers. Chemistry 2022, 28, e202104442. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Wang, Z.; Liu, X.; Jiang, Y.; Jiao, X.; Xie, P. Total Synthesis of the Proposed Structure of Characellide B. Org. Lett. 2021, 23, 3680–3684. [Google Scholar] [CrossRef]
- Zhang, H.H.; Wang, C.S.; Li, C.; Mei, G.J.; Li, Y.; Shi, F. Design and Enantioselective Construction of Axially Chiral Naphthyl-Indole Skeletons. Angew. Chem. Int. Ed. 2017, 56, 116–121. [Google Scholar] [CrossRef]
- Wang, Y.B.; Tan, B. Construction of Axially Chiral Compounds via Asymmetric Organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. [Google Scholar] [CrossRef]
- Chen, K.W.; Chen, Z.H.; Yang, S.; Wu, S.F.; Zhang, Y.C.; Shi, F. Organocatalytic Atroposelective Synthesis of N-N Axially Chiral Indoles and Pyrroles by De Novo Ring Formation. Angew. Chem. Int. Ed. 2022, 61, e202116829. [Google Scholar]
- Chen, Y.H.; Li, H.H.; Zhang, X.; Xiang, S.H.; Li, S.; Tan, B. Organocatalytic Enantioselective Synthesis of Atropisomeric Aryl-p-Quinones: Platform Molecules for Diversity-Oriented Synthesis of Biaryldiols. Angew. Chem. Int. Ed. 2020, 59, 11374–11378. [Google Scholar] [CrossRef]
- Yang, G.-H.; Zheng, H.; Li, X.; Cheng, J.-P. Asymmetric Synthesis of Axially Chiral Phosphamides via Atroposelective N-Allylic Alkylation. ACS Catal. 2020, 10, 2324–2333. [Google Scholar] [CrossRef]
- Kitagawa, O. A unique method for highly enantioselective synthesis of N–C axially chiral compounds. Chem 2021, 7, 1696–1698. [Google Scholar] [CrossRef]
- Liu, Z.-S.; Xie, P.-P.; Hua, Y.; Wu, C.; Ma, Y.; Chen, J.; Cheng, H.-G.; Hong, X.; Zhou, Q. An axial-to-axial chirality transfer strategy for atroposelective construction of C–N axial chirality. Chem 2021, 7, 1917–1932. [Google Scholar] [CrossRef]
- Yang, B.; Yang, J.; Zhang, J. Synthesis of Axially Chiral Anilides Enabled by a Palladium/ Ming-Phos-Catalyzed Desymmetric Sonogashira Reaction. Chin. J. Chem. 2021, 40, 317–322. [Google Scholar] [CrossRef]
- Gan, K.B.; Zhong, R.L.; Zhang, Z.W.; Kwong, F.Y. Atropisomeric Phosphine Ligands Bearing C-N Axial Chirality: Applications in Enantioselective Suzuki-Miyaura Cross-Coupling Towards the Assembly of Tetra-ortho-Substituted Biaryls. J. Am. Chem. Soc. 2022, 144, 14864–14873. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chen, B.; Yao, Y.P.; Zhou, H.J.; Wang, X.; Wang, N.Z.; Chen, F.E. Construction of Non-Biaryl Atropisomeric Amide Scaffolds Bearing a C-N Axis via Enantioselective Catalysis. Molecules 2022, 27, 6583. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, N.; Zhong, S.; Wen, Z.; Wang, T. A Central-to-Axial Chirality Conversion Strategy for the Synthesis of C-N Axially Chiral N-Arylpyrroles. Org. Lett. 2022, 24, 2842–2846. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Jiang, F.; Shi, F. Organocatalytic Asymmetric Synthesis of Indole-Based Chiral Heterocycles: Strategies, Reactions, and Outreach. Acc. Chem. Res. 2020, 53, 425–446. [Google Scholar] [CrossRef]
- Ma, C.; Jiang, F.; Sheng, F.T.; Jiao, Y.; Mei, G.J.; Shi, F. Design and Catalytic Asymmetric Construction of Axially Chiral 3,3′-Bisindole Skeletons. Angew. Chem. Int. Ed. 2019, 58, 3014–3020. [Google Scholar] [CrossRef]
- Hang, Q.-Q.; Wu, S.-F.; Yang, S.; Wang, X.; Zhong, Z.; Zhang, Y.-C.; Shi, F. Design and catalytic atroposelective synthesis of axially chiral isochromenone-indoles. Sci. China. Chem. 2022, 65, 1929–1937. [Google Scholar] [CrossRef]
- Xia, W.; An, Q.J.; Xiang, S.H.; Li, S.; Wang, Y.B.; Tan, B. Chiral Phosphoric Acid Catalyzed Atroposelective C-H Amination of Arenes. Angew. Chem. Int. Ed. 2020, 59, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Guo, J.; Liu, J.; Cao, W.; Wei, C.; Zhang, Y.; Zhang, X.; Fu, Z. Organocatalytic dynamic kinetic resolution of N-arylindole lactams: Atroposelective construction of axially chiral amino acids bearing a C-N chiral axis. Sci. China Chem. 2022, 65, 905–911. [Google Scholar] [CrossRef]
- Wang, G.; Huang, J.; Zhang, J.; Fu, Z. Catalytically atroposelective ring-opening of configurationally labile compounds to access axially chiral biaryls. Org. Chem. Front. 2022, 9, 4507–4521. [Google Scholar] [CrossRef]
- Qin, W.; Liu, Y.; Yan, H. Enantioselective Synthesis of Atropisomers via Vinylidene ortho-Quinone Methides (VQMs). Acc. Chem. Res. 2022, 55, 2780–2795. [Google Scholar] [CrossRef]
- Liang, H.; Zhu, G.; Pu, X.; Qiu, L. Copper-Catalyzed Enantioselective C-H Arylation between 2-Arylindoles and Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9246–9250. [Google Scholar] [CrossRef]
- Wang, C.S.; Wei, L.; Fu, C.; Wang, X.H.; Wang, C.J. Asymmetric Synthesis of Axially Chiral Naphthyl-C3-indoles via a Palladium-Catalyzed Cacchi Reaction. Org. Lett. 2021, 23, 7401–7406. [Google Scholar] [CrossRef]
- Corti, V.; Thogersen, M.K.; Enemaerke, V.J.; Rezayee, N.M.; Barlose, C.L.; Anker Jorgensen, K. Construction of C-N Atropisomers by Aminocatalytic Enantioselective Addition of Indole-2-carboxaldehydes to o-Quinone Derivatives. Chemistry 2022, e202202395. [Google Scholar] [CrossRef]
- Liu, J.; Li, Q.; Shao, Y.; Sun, J. Atroposelective Synthesis of Axially Chiral C2-Arylindoles via Rhodium-Catalyzed Asymmetric C-H Bond Insertion. Org. Lett. 2022, 24, 4670–4674. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Shan, W.; Liao, R.; Deng, Y.; Peng, F.; Shao, Z. Construction of Axially Chiral Indoles by Cycloaddition–Isomerization via Atroposelective Phosphoric Acid and Silver Sequential Catalysis. ACS Catal. 2022, 12, 8094–8103. [Google Scholar] [CrossRef]
- Xu, M.M.; Cao, W.B.; Xu, X.P.; Ji, S.J. Visible-Light-Promoted Radical Cyclization and N−N Bond Cleavage Relay of N-Aminopyridinium Ylides for Access to 2,3-Difunctionalized Indoles. Adv. Synth. Catal. 2022, 364, 2211–2220. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, P.; Kong, L.; Li, X. Rhodium-Catalyzed Atroposelective C-H Arylation of (Hetero)Arenes Using Carbene Precursors as Arylating Reagents. Org. Lett. 2022, 24, 3189–3193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Q.; Wu, J.; Fan, J.; Xie, M. Construction of N-C Axial Chirality through Atroposelective C-H Olefination of N-Arylindoles by Palladium/Amino Acid Cooperative Catalysis. Org. Lett. 2019, 21, 6361–6365. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J.; Chen, J.J.; Feng, C.F.; Li, H.Y.; Wang, X.; Xu, H.; Dai, H.X. Synthesis of Axially Chiral Olefin-Oxazoline Ligands via Pd-Catalyzed Multiple C-H Functionalization. Org. Lett. 2020, 22, 9169–9173. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liou, Y.C.; Chen, X.; Ackermann, L. Thioether-enabled palladium-catalyzed atroposelective C-H olefination for N-C and C-C axial chirality. Chem. Sci. 2022, 13, 4088–4094. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, Y.; Jing, J.; Wang, F.; Li, X. Rhodium(III)-Catalyzed Atroposelective Synthesis of C-N Axially Chiral Naphthylamines and Variants via C-H Activation. Org. Lett. 2022, 24, 2531–2535. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, X.; Min, X.L.; Yi, W.; Liu, P.; He, Y. Tandem Iridium Catalysis as a General Strategy for Atroposelective Construction of Axially Chiral Styrenes. J. Am. Chem. Soc. 2021, 143, 10686–10694. [Google Scholar] [CrossRef]
- Wu, Y.-X.; Liu, Q.; Zhang, Q.; Ye, Z.; He, Y. Asymmetric allylic substitution-isomerization for accessing axially chiral vinylindoles by intramolecular π … π stacking interactions. Cell Rep. Physic. Sci. 2022, 3, 101005. [Google Scholar] [CrossRef]
- Zhang, X.L.; Gu, J.; Cui, W.H.; Ye, Z.; Yi, W.; Zhang, Q.; He, Y. Stepwise Asymmetric Allylic Substitution-Isomerization Enabled Mimetic Synthesis of Axially Chiral B,N-Heterocycles. Angew. Chem. Int. Ed. 2022, e202210456. [Google Scholar] [CrossRef]
- Li, Z.; Tang, M.; Hu, C.; Yu, S. Atroposelective Haloamidation of Indoles with Amino Acid Derivatives and Hypohalides. Org. Lett. 2019, 21, 8819–8823. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Yu, S. NaClO-Promoted Atroposelective Couplings of 3-Substituted Indoles with Amino Acid Derivatives. Org. Lett. 2019, 21, 4754–4758. [Google Scholar] [CrossRef]
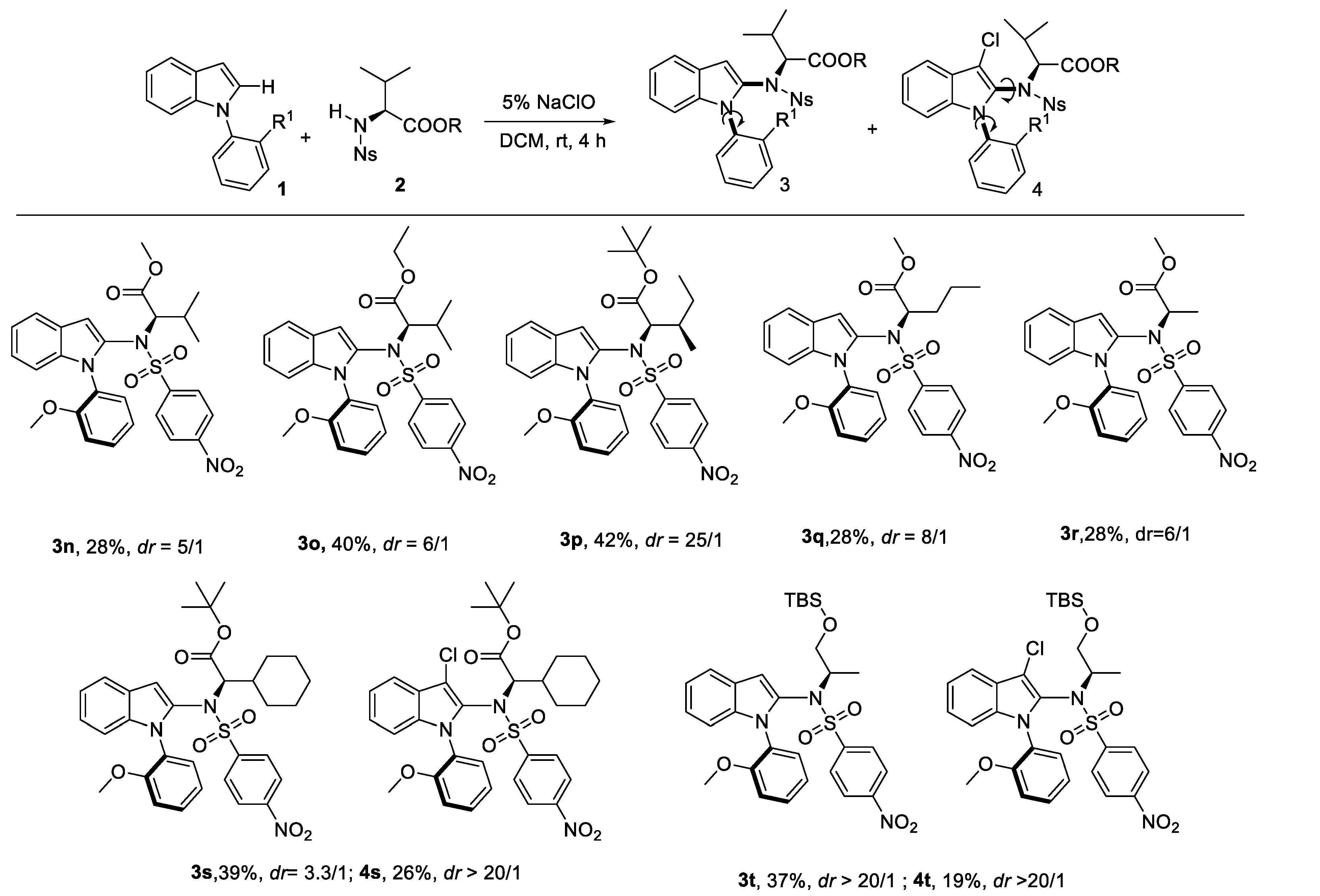
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).