Abstract
Glycerol is the main side product in the biodiesel manufacturing process, and the development of glycerol valorization methods would indirectly contribute the sustainable biodiesel production and decarbonization. Transformation of glycerol to optically active C3 units would be one of the attractive routes for glycerol valorization. We herein present the asymmetric sulfonylative desymmetrization of glycerol by using a CuCN/(R,R)-PhBOX catalyst system to provide an optically active monosulfonylated glycerol in high efficiency. A high degree of enantioselectivity was achieved with a commercially available chiral ligand and an inexpensive carbonate base. The optically active monosulfonylated glycerol was successfully transformed into a C3 unit attached with differentially protected three hydroxy moieties. In addition, the synthetic utility of the present reaction was also demonstrated by the transformation of the monosulfonylated glycerol into an optically active synthetic ceramide, sphingolipid E.
1. Introduction
The use of renewable energies instead of conventional fossil fuels has become a global trend from the perspective of expected fossil fuel depletion and global climate change. Biodiesel fuel, which is produced by the transesterification of vegetable or animal fats with methanol, has emerged as a promising alternative to petroleum-derived diesel fuel [1,2]. The biodiesel manufacturing process inevitably provides 10 wt% of glycerol (1,2,3-propanetriol) as the main side product, and an oversupply of the crude glycerol would be projected with the growing biodiesel market [3]. Thus, great effort has been devoted to the development of an efficient process for the transformation of glycerol to value-added commodity chemicals, which would indirectly contribute to sustainable biodiesel production [4,5,6,7]. Among them, optically active glycerol derivatives would be an attractive target in the field of glycerol valorization. Chiral glycerol derivatives such as glyceraldehyde and glycidyl tosylate are utilized as valuable C3 building blocks in medicinal [8,9,10,11,12] and synthetic organic chemistry [13,14,15,16,17,18]. A chiral pool approach is a traditional strategy to access enantiopure glycerol derivatives, but the need for multi-step transformations may be a major drawback [19,20,21,22,23]. Asymmetric desymmetrization of glycerol would be one of the most straightforward methods for chiral glycerol derivatives production. Several types of enzymes, i.e., lipase, kinase, and dehydrogenase/oxidase, have been successfully applied to this strategy, affording optically active glycerols with various enantioselectivities [24,25,26,27,28,29]. On the other hand, despite the recent development of the enantioselective desymmetrization [30,31] of 1,2-diols [32,33,34,35,36,37,38,39] and 1,3-diols [40,41,42,43,44,45,46,47,48], including C2-substituted glycerols [49,50,51,52,53], the non-enzymatic direct desymmetrization of glycerol is still a challenging task presumably due to an extremely high hydrophilic nature of glycerol. In this context, the use of 2-O-protected glycerol derivatives would be the most common strategy for the chemical desymmetrization of glycerol (Scheme 1a) [54,55,56,57]. In 2013, Tan et al. developed the first non-enzymatic direct desymmetrization of glycerol through organocatalyzed enantioselective silylation (Scheme 1b) [58]. In their protocol, the high enantioselectivity was achieved through the secondary kinetic resolution on the initially formed monosilylated glycerol. Very recently, the copper-catalyzed sulfonylative desymmetrization of glycerol using a non-commercially available ligand with silver carbonate was described in the Chinese patent [59]. Although the desired product was obtained with high enantioselectivity under copper-catalyzed conditions, the use of a commercially available chiral ligand and a non-precious metal base would be desirable from a practical and economical point of view [60,61,62]. Herein, we report the asymmetric desymmetrization of glycerol through sulfonylation with a Cu/(R,R)-PhBOX complex and sodium carbonate affording the optically active monosulfonylated glycerol in an excellent yield and enantioselectivity (Scheme 1c) [63].
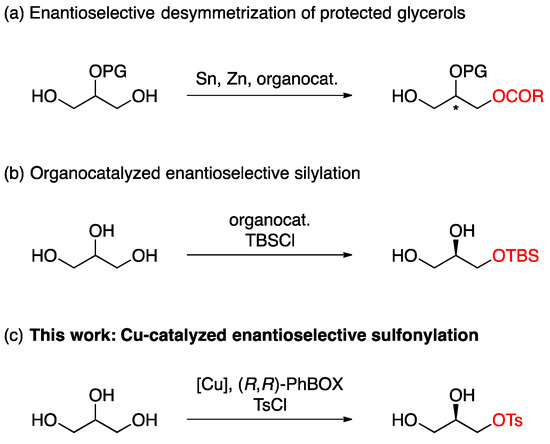
Scheme 1.
Asymmetric desymmetrization of glycerols. (a) Enantioselective desymmetrization of protected glycerols. (b) Organocatalyzed enantioselective silylation. (c) This work: Cu-catalyzed enantioselective sulfonylation. The asterisk denotes the chiral center.
2. Results and Discussion
For the initial attempt to optimize the enantioselective desymmetrization of glycerol, compound 1 was treated with p-toluenesulfonyl chloride (TsCl) in the presence of copper trifluoromethanesulfonate (Cu(OTf)2)/(R,R)-PhBOX and sodium carbonate in acetonitrile. Pleasingly, the desired monotosylated glycerol 2 was obtained in 91% yield with 83% ee (Table 1, entry 1). Using other carbonate salts, i.e., potassium carbonate and cesium carbonate, resulted in a decrease in both yield and enantioselectivity (entries 2 and 3). Organic bases were not suitable for the present transformation (entries 4 and 5). Next, other copper catalysts were examined to evaluate the catalytic activity in this reaction system. While CuCl provided 2 with a slightly lowered yield and enantioselectivity, CuBr and CuI exhibited a similar reactivity compared with Cu(OTf)2 (entries 6–8). The use of CuCN led to the formation of 2 in 83% yield with higher enantioselectivity, and the reaction concentration was able to be doubled without significant changes regarding both yield and enantioselectivity (entries 9–10). Pleasingly, we found that acetone was a better solvent choice to afford the desired product in an excellent yield and enantioselectivity (96% yield, 94% ee), and the concentration of 0.25 M was found to be suitable for the present reaction (entries 11–12). The catalyst loading was able to be reduced to 5 mol% without a significant decrease in the yield and enantioselectivity (entry 13). We also examined the feasibility of the gram-scale preparation of 2. The reaction with 6.0 mmol of glycerol successfully provided the desired product 2 in 88% yield (1.30 g) with 93% ee (entry 14). Control experiments revealed that both the copper salt and the BOX ligand were essential to promote the tosylation of 1 (entries 15–16).

Table 1.
Optimization of reaction conditions 1.
In order to gain insight into the chemoselectivity of the present reaction, we performed competition studies with alcohol additives (Table 2). The addition of n-propanol (1.0 eq) led to a slight decrease in both yield and enantioselectivity, but the formation of n-propyl tosylate was not detected (entry 1 vs. entry 2). Moreover, selective sulfonylation of glycerol (1) over 1,2- and 1,3-diols was observed under the present reaction conditions, and the desired monosulfonylated glycerol 2 was obtained without a significant loss of enantioselectivity (entry 1 vs. entries 3–4). In addition, the reaction of 2-O-benzylglycerol (4) provided the corresponding monotosylated product 5 in a low yield with poor enantioselectivity (Scheme 2). These results indicated that the present reaction system would be highly selective for the glycerol transformation even in the presence of other alcohols, and the presence of a free 2-hydroxy moiety would play a crucial role in accelerating the tosylation with high asymmetric induction.
Table 2.
Competition studies 1.
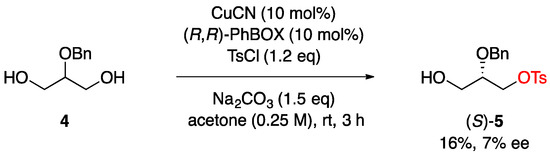
Scheme 2.
Enantioselective desymmetrization of 2-O-benzylglycerol.
With successful asymmetric desymmetrization of glycerol achieved, we then investigated the synthetic applications of the obtained optically active glycerol. First, the site-selective protection of the remained hydroxy groups in (R)-2 was examined (Scheme 3). The primary hydroxy moiety was selectively protected by using tert-butyldimethylsilyl chloride (TBSCl) with imidazole, affording the corresponding product (R)-6. Acetylation of the secondary hydroxy group with Ac2O in the presence of a DMAP catalyst provided (R)-7 in an excellent yield. Since each protective group would be removed by different deprotecting protocols, (R)-7 would be potentially useful as a versatile chiral C3 building block.
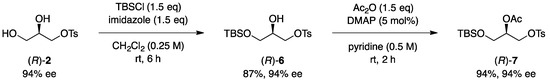
Scheme 3.
Synthesis of optically active glycerol derivatives.
Next, we turned our attention to the application of the present transformation in the synthesis of an optically active synthetic ceramide. Ceramides are major components of the lamellar structure in stratum corneum lipids which protect the epidermis from excess transepidermal water loss and from the permeation of pathogens [64]. Interestingly, optically active natural ceramides showed different thermotropic behavior from racemic variants, and the lamellar liquid crystalline system containing optically active natural ceramides improved recovering effects of a water-holding ability and a barrier function of the skin [65]. Sphingolipid E (SLE) was a synthetic ceramide designed and synthesized by Kao Corporation as a structural analog of natural type 2 ceramide and was found to form a stable lamellar structure that exhibits a high water-holding ability [66]. Although SLE has been utilized as a racemic form, optically active SLE might affect the physicochemical property to form a lamellar structure and the interaction mode with water molecules. The synthesis of optically active SLE commenced with the introduction of nitrogen functionality to (R)-2 (Scheme 4). The nucleophilic azide substitution of the tosyloxy group in (R)-2 with sodium azide provided azide diol (S)-8 in an excellent yield in the presence of 15-crown-5. The enantiomeric excess of (S)-8 was determined after the tosylation of the primary hydroxy group, and no obvious racemization was observed in the azide substitution step. The boronic acid-catalyzed site-selective alkylation [67] of (S)-8 with cetyl bromide followed by the Pd/C-catalyzed reduction of the azide group afforded the corresponding aminoalcohol (S)-10. N-Alkylated product (S)-11 was successfully obtained by the reductive amination of (S)-10 with TBS-protected glycolaldehyde using 2-picoline borane as a reductant. Finally, (S)-11 was transformed into the optically active synthetic ceramide (S)-12 via the amidation with palmitoyl chloride followed by the removal of the TBS group.
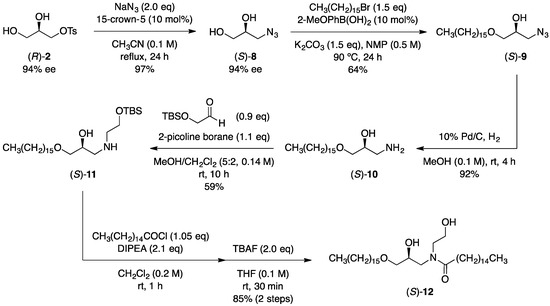
Scheme 4.
Synthesis of an optically active synthetic ceramide.
In conclusion, we have developed the copper-catalyzed asymmetric sulfonylative desymmetrization of glycerol. The reaction smoothly proceeded under mild reaction conditions with a commercially available (R,R)-PhBOX ligand and an inexpensive inorganic base, providing the optically active monotosylated glycerol derivative in a high yield with high enantiomeric excess. The synthetic utility of the present transformation was demonstrated by the preparation of an enantio-enriched C3 building block with three different types of protective groups. Moreover, the synthesis of the optically active synthetic ceramide was also achieved from the monotosylated glycerol in six steps without a notable loss of enantiopurity.
3. Materials and Methods
3.1. General
Unless otherwise noted, all reactions were performed under an argon atmosphere, and all reagents and solvents were used as received without further purification. Column chromatography was performed on Fuji silysia Chromatorex 60B silica gel. Melting points (mp) were measured with a Yanako Micro Melting Point Apparatus MP-J3 and reported without correction. Infrared (IR) spectra were recorded on a Shimadzu IRAffinity-1 spectrometer and expressed as frequency of absorption (cm−1). Optical rotations were measured with JASCO DIP-1000 or P-2200 spectrometers. 1H and 13C{1H} NMR spectra (Supplementary Materials) were recorded on JEOL JNM-AL400, JNM-ECZ400R (400 MHz for 1H NMR, 100 MHz for 13C{1H} NMR) or Varian NMR System 500PS SN (125 MHz for 13C{1H} NMR) spectrometers. Chemical shift values are expressed in parts per million (ppm) relative to internal TMS (δ 0.00 ppm for 1H NMR) or deuterated solvent peaks (δ 77.0 ppm (CDCl3) for 13C{1H} NMR). Abbreviations are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; app, apparent. Enantiomeric excess values were determined by chiral high-performance liquid chromatography (HPLC) analysis using DAICEL CHIRALPAK AD or AY-H columns. HPLC chromatograms (Supplementary Materials) were recorded on a CR8A CHROMATOPAC with an LC-20AT pump and SPD-20A UV detector (Shimadzu). High-resolution mass spectra (HRMS) were obtained on a JEOL JMS-700N (double-focusing magnetic sector mass analyzer) spectrometer with either the electron impact ionization (EI) or the fast atom bombardment (FAB) methods or a JEOL JMS-T100TD (TOF mass analyzer) spectrometer with either the direct analysis in real-time (DART) or the electrospray ionization (ESI) method.
3.2. Copper-Catalyzed Asymmetric Desymmetrization of Glycerol
A solution of (R,R)-PhBOX (33.4 mg, 0.10 mmol) and CuCN (8.96 mg, 0.10 mmol) in CH2Cl2 (4.0 mL) was stirred for 3 h at 40 °C. The reaction mixture was allowed to cool to room temperature and filtered into a round bottom flask using a cotton plug. After removal of the solvent under reduced pressure, the resulting solid was dried in vacuo for 30 min. To the round-bottom flask containing CuCN/(R,R)-PhBOX was added a solution of glycerol (92.1 mg, 1.0 mmol) in acetone (4.0 mL), and the resulting mixture was stirred for 10 min at room temperature. To the mixture was successively added Na2CO3 (159 mg, 1.5 mmol) and TsCl (229 mg, 1.2 mmol), and the mixture was stirred for 3 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl, and the resulting mixture was extracted with AcOEt. Combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/AcOEt = 1/2) to afford (R)-2 (236 mg, 0.96 mmol, 96% yield).
(R)-2,3-Dihydroxypropyl 4-methylbenzenesulfonate ((R)-2). White solid; mp = 56–58 °C; [α]D23 −8.3 (c 1.00, MeOH) for 94% ee; 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.5 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 4.13–4.06 (m, 2H), 3.99–3.93 (m, 1H), 3.74–3.69 (m, 1H), 3.66–3.60 (m, 1H), 2.51 (d, J = 5.5 Hz, 1H), 2.46 (s, 3H), 1.94 (t, J = 5.9 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 145.2, 132.2, 130.0, 127.9, 70.7, 69.6, 62.7, 21.6; IR (ATR): 3368, 2926, 1350, 1171, 968, 812 cm−1; HRMS (EI) m/z: [M]+ calcd for C10H14O5S 246.0562, found 254.0562; HPLC analysis: Chiralpak AY-H, hexane/EtOH = 2/1, flow rate 1.0 mL/min, wavelength 254 nm, tR 13.5 min (minor) and 15.6 min (major). The absolute configuration of 2 was established by comparing the sign of the specific rotation of 2 with the literature value ([α]D22 −9.3 (c 4.99, MeOH) for (R)-2) [68].
3.3. Large-Scale Experiment
A solution of (R,R)-PhBOX (200 mg, 0.60 mmol) and CuCN (53.7 mg, 0.60 mmol) in CH2Cl2 (24 mL) was stirred for 3 h at 40 °C. The reaction mixture was allowed to cool to room temperature and filtered into a round bottom flask using a cotton plug. After removal of the solvent under reduced pressure, the resulting solid was dried in vacuo for 1 h. To the round-bottom flask containing CuCN/(R,R)-PhBOX was added a solution of glycerol (553 mg, 6.0 mmol) in acetone (24 mL), and the resulting mixture was stirred for 10 min at room temperature. To the mixture was successively added Na2CO3 (950 mg, 9.0 mmol) and TsCl (1.37 g, 7.2 mmol), and the mixture was stirred for 9 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl, and the resulting mixture was extracted with AcOEt. Combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/AcOEt = 1/2) to afford (R)-2 (1.30 g, 5.28 mmol, 88% yield, 93% ee).
3.4. Copper-Catalyzed Asymmetric Desymmetrization of 2-O-Benzylglycerol
The reaction was performed using 2-O-benzylglycerol (4, 182 mg, 1.0 mmol) [69] according to the procedure described in Section 3.2. Silica gel column chromatography (hexane/acetone = 3/1) to afford (S)-5 (52.6 mg, 0.16 mmol, 16% yield).
(S)-2-(Benzyloxy)-3-hydroxypropyl 4-methylbenzenesulfonate ((S)-5). colorless oil; [α]D21 −2.1 (c 1.00, CHCl3) for 7% ee; 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 8.2 Hz, 2H), 7.36–7.27 (m, 7H), 4.63 (d, J = 11.7 Hz, 1H), 4.55 (d, J = 11.7 Hz, 1H), 4.17–4.10 (m, 2H), 3.75–3.67 (m, 2H), 3.62–3.56 (m, 1H), 2.45 (s, 3H), 1.80 (t, J = 6.3 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3): δ 145.0, 137.5, 132.6, 129.9, 128.5, 128.1, 128.0, 127.9, 76.7, 72.5, 68.7, 61.4, 21.7; IR (ATR): 3445, 3032, 2879, 1597, 1354, 1173, 974 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C17H20NaO5S 359.0929, found 359.0922; HPLC analysis: Chiralpak AD, hexane/EtOH = 3/1, flow rate 1.0 mL/min, wavelength 254 nm, tR 7.1 min (minor) and 8.8 min (major). The absolute configuration of 5 was established by comparing the sign of the specific rotation of 5 with the literature value ([α]D22 +29.5 (c 1.01, CHCl3) for (R)-5) [70].
3.5. Synthesis of Optically Active Glycerol Derivatives
(R)-3-((tert-Butyldimethylsilyl)oxy)-2-hydroxypropyl 4-methylbenzenesulfonate ((R)-6). To a solution of (R)-2 (134 mg, 0.54 mmol) in CH2Cl2 (2.2 mL) was successively added imidazole (55.8 mg, 0.82 mmol) and TBSCl (123 mg, 0.82 mmol) at room temperature. After stirring for 6 h at the same temperature, the reaction was quenched with H2O. The resulting mixture was extracted with AcOEt. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/AcOEt = 2/1) to afford (R)-6 (103 mg, 0.287 mmol, 87% yield) as a colorless oil; [α]D27 −12.4 (c 1.01, AcOEt) for 94% ee; 1H NMR (400 MHz, CDCl3): δ 7.82–7.79 (m, 2H), 7.37–7.34 (m, 2H), 4.07 (dd, J = 9.9, 5.6 Hz, 1H), 4.01 (dd, J = 9.9, 5.6 Hz, 1H), 3.87–3.83 (m, 1H), 3.66–3.59 (m, 2H), 2.45 (s, 3H), 2.40 (d, J = 6.2 Hz, 1H), 0.85 (s, 9H), 0.039 (s, 3H), 0.035 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 145.0, 132.6, 129.9, 128.0, 70.0, 69.2, 62.8, 25.7, 21.6, 18.1, −5.6; IR (ATR): 3537, 2930, 2857, 1360, 1252, 1175, 980, 833 cm−1; HRMS (EI) m/z: [M]+ calcd for C16H28O5SSi 360.1427, found 360.1429. HPLC analysis: Chiralpak AY-H, hexane/i-PrOH = 10/1, flow rate 1.0 mL/min, wavelength 254 nm, tR 22.4 min (minor) and 25.3 min (major).
(R)-1-((tert-Butyldimethylsilyl)oxy)-3-(tosyloxy)propan-2-yl acetate ((R)-7). To a solution of (R)-6 (144 mg, 0.40 mmol) in pyridine (0.80 mL) was successively added DMAP (2.44 mg, 0.020 mmol) and Ac2O (49.0 mg, 0.48 mmol) at room temperature. After stirring for 2 h at the same temperature, the reaction mixture was diluted with toluene (5.0 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/AcOEt = 5/1) to afford (R)-7 (151 mg, 0.378 mmol, 94% yield) as a colorless oil. [α]D27 −2.2 (c 1.24, AcOEt) for 94% ee; 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.96–4.91 (m, 1H), 4.22 (dd, J = 10.7, 3.7 Hz, 1H), 4.17 (dd, J = 10.9, 5.4 Hz, 1H), 3.70–3.63 (m, 2H), 2.45 (s, 3H), 2.00 (s, 3H), 0.83 (s, 9H), 0.01 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3): δ 170.0, 144.9, 132.7, 129.8, 128.0, 71.3, 67.8, 60.5, 25.6, 21.6, 20.8, 18.1, −5.58, −5.62; IR (ATR): 1744, 1362, 1233, 1175, 988, 833 cm−1; HRMS (EI) m/z: [M]+ calcd for C18H30O6SSi 402.1532, found 402.1533; HPLC analysis: Chiralpak AD, hexane/EtOH = 20/1, flow rate 1.0 mL/min, wavelength 254 nm, tR 4.8 min (major) and 6.5 min (minor).
3.6. Synthesis of an Optically Active Synthetic Ceramide
(S)-3-Azidopropane-1,2-diol ((S)-8). To a solution of (R)-2 (246 mg, 1.0 mmol) in CH3CN (10 mL) was successively added 15-crown-5 (22.1 mg, 0.10 mmol) and NaN3 (130 mg, 2.0 mmol) at room temperature. After refluxing for 24 h, the reaction mixture was filtered using celite, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 12/1) to afford (S)-8 (114 mg, 0.974 mmol, 97% yield) as a colorless oil; [α]D23 −16.2 (c 1.10, MeOH); 1H NMR (400 MHz, CDCl3): δ 3.92–3.86 (m, 1H), 3.74–3.72 (m, 1H), 3.65–3.61 (m, 1H), 3.47–3.38 (m, 2H), 2.49 (d, J = 4.1 Hz, 1H), 1.95 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 70.9, 63.9, 53.4; IR (ATR): 3333, 2924, 2855, 2093, 1443, 1272, 1103, 1038, 928 cm−1; HRMS (EI) m/z: [M]+ calcd for C3H7N3O2 117.0538, found 117.0546. The absolute configuration of 8 was established by comparing the sign of the specific rotation of 8 with the literature value ([α]D20 −17.4 (c 1.00, MeOH) for (S)-8) [71].
(S)-1-Azido-3-(hexadecyloxy)propan-2-ol ((S)-9). To a solution of (S)-8 (46.8 mg, 0.40 mmol) in N-methylpyrrolidone (0.80 mL) was successively added 2-methoxyphenylboronic acid (6.0 mg, 0.040 mmol) and K2CO3 (82.9 mg, 0.60 mmol). The resulting mixture was stirred for 30 min at room temperature, and then cetyl bromide (183 mg, 0.60 mmol) was added. After stirring for 24 h at 95 °C, the reaction mixture was diluted with H2O and extracted with AcOEt. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/Et2O = 8/2) to afford (S)-9 (87.3 mg, 0.256 mmol, 64% yield) as a white solid; mp = 37–39 °C; [α]D24 −11.8 (c 1.00, MeOH); 1H NMR (400 MHz, CDCl3): δ 3.97–3.91 (m, 1H), 3.51–3.33 (m, 6H), 2.42 (d, J = 5.0 Hz, 1H), 1.61–1.54 (m, 2H), 1.32–1.26 (m, 26H), 0.88 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 71.8, 71.7, 69.6, 53.5, 31.9, 29.68, 29.66, 29.64, 29.59, 29.57, 29.5, 29.4, 29.3, 26.1, 22.7, 14.1; IR (ATR): 3429, 2914, 2876, 2846, 2088, 1466, 1337, 1290, 1113, 989 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C19H40N3O2 342.3121, found 342.3171.
(S)-1-Amino-3-(hexadecyloxy)propan-2-ol ((S)-10). To a reaction vessel charged with 10% Pd/C (24.9 mg, 10% w/w) was added a solution of (S)-9 (249 mg, 0.73 mmol) in MeOH (7.3 mL) at room temperature under argon atmosphere. The reaction vessel was charged with H2 gas, and then the mixture was stirred for 4 h at room temperature. The reaction mixture was filtered using celite, and then the filtrate was concentrated under reduced pressure. The residue was dissolved in 10% aqueous HCl and washed with AcOEt. The aqueous layer was basified with saturated aqueous NaHCO3 and then extracted with CHCl3. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to afford (S)-10 (211 mg, 0.67 mmol, 92% yield) as a white solid; mp = 60–61 °C; [α]D24 −3.2 (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ 3.76–3.70 (m, 1H), 3.50–3.43 (m, 3H), 3.38 (dd, J = 9.5, 6.5 Hz, 1H), 2.83 (dd, J = 12.7, 3.1 Hz, 1H), 2.72 (dd, J = 12.5, 6.7 Hz, 1H), 1.61–1.54 (m, 2H), 1.32–1.26 (m, 26H), 0.88 (t, J = 6.7 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 73.0, 71.7, 71.1, 44.4, 31.9, 29.7, 29.64, 29.59, 29.58, 29.5, 29.3, 26.1, 22.7, 14.1; IR (ATR): 2912, 2827, 1470, 1130, 1032, 924 cm−1; HRMS (FAB) m/z: [M + H]+ calcd for C19H42NO2 316.3216, found 316.3200.
(S)-2,2,3,3-Tetramethyl-4,11-dioxa-7-aza-3-silaheptacosan-9-ol ((S)-11). To a solution of 2-(tert-butyldimethylsilyloxy)acetaldehyde (34.9 mg, 0.20 mmol) [72] in MeOH/CH2Cl2 (5:2, 1.4 mL) was added (S)-10 (69.4 mg, 0.22 mmol) at room temperature. After stirring for 10 min at the same temperature, 2-picoline borane (256 mg, 0.24 mmol) was added, and then the reaction mixture was stirred for an additional 10 h. The reaction mixture was diluted with H2O and extracted with CH2Cl2. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 12/1) to afford (S)-11 (55.8 mg, 0.117 mmol, 59% yield) as a colorless amorphous; [α]D25 −3.6 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3): δ 3.86–3.81 (m, 1H), 3.75–3.67 (m, 2H), 3.49–3.39 (m, 4H), 2.78–2.64 (m, 4H), 1.60–1.54 (m, 2H), 1.33–1.25 (m, 26H), 0.90–0.86 (m, 12H), 0.06 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3): δ 73.3, 71.7, 68.7, 62.2, 51.7, 51.5, 31.9, 29.7, 29.62, 29.58, 29.5, 29.3, 26.1, 25.9, 22.7, 18.3, 14.1, −5.4; IR (ATR): 2914, 2849, 1472, 1464, 1256, 1119, 1080, 968, 937, 831 cm−1; HRMS (FAB) m/z: [M + H]+ calcd for C27H60NO3Si 473.4342, found 473.4300.
(S)-N-(3-(Hexadecyloxy)-2-hydroxypropyl)-N-(2-hydroxyethyl)palmitamide ((S)-12). To a solution of (S)-11 (135 mg, 0.28 mmol) and i-Pr2NEt (77.0 mg, 0.60 mmol) in CH2Cl2 (1.4 mL) was added palmitoyl chloride (81.9 mg, 0.30 mmol) at room temperature. After stirring for 1 h at the same temperature, all volatile was removed under reduced pressure. The residue was dissolved in THF (2.8 mL), and then a 1.0 M solution of TBAF in THF (0.57 mL) was added at room temperature. After stirring for 30 min at the same temperature, the reaction mixture was diluted with H2O and extracted with CHCl3. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/AcOEt = 1/1) to afford (S)-12 (144 mg, 0.241 mmol, 85% yield) as a white solid; mp = 67–68 °C; [α]D25 −4.4 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3, mixture of rotamers): δ 4.16–3.93 (m, 1H), 3.85–3.74 (m, 2H), 3.67–3.25 (m, 8H), 2.46–2.30 (m, 2H), 1.67–1.52 (m, 4H), 1.28 (m, 50H), 0.88 (app t, J = 6.9 Hz, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 175.8, 72.4, 72.1, 71.8, 71.6, 69.8, 69.4, 61.7, 60.6, 53.3, 52.5, 51.4, 51.1, 33.6, 33.5, 31.9, 29.7, 29.62, 29.60, 29.59, 29.56, 29.53, 29.47, 29.44, 29.42, 29.3, 26.1, 26.0, 25.29, 25.26, 22.7, 14.1; IR (ATR): 3320, 2916, 2849, 1611, 1464, 1437, 1375, 1306, 1290, 1261, 1206, 1165, 1109, 1094, 1059, 1040, 955, 845, 814 cm−1; HRMS (FAB) m/z: [M + H]+ calcd for C37H76NO4 598.5773, found 598.5800.
3.7. Tosylation of Azide Diol (S)-8
(S)-3-Azido-2-hydroxypropyl 4-methylbenzenesulfonate ((S)-8′). To a solution of (S)-8 (23.4 mg, 0.20 mmol) in CH3CN (0.80 mL) was added pyridine (23.7 mg, 0.30 mmol) and TsCl (57.2 mg, 0.30 mmol) at room temperature. After stirring for 10 h at the same temperature, the reaction was quenched with H2O, and the resulting mixture was extracted with AcOEt. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/AcOEt = 2/1) to afford (S)-8′ (30.3 mg, 0.112 mmol, 56% yield) as a colorless oil; [α]D27 −17.5 (c 1.00, AcOEt) for 94% ee; 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.3 Hz, 2H), 4.09–3.98 (m, 3H), 3.43 (dd, J = 12.9, 4.6 Hz, 1H), 3.38 (dd, J = 12.7, 5.4 Hz, 1H), 2.47 (s, 3H), 2.40 (d, J = 5.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 145.4, 132.3, 130.0, 128.0, 70.5, 68.5, 52.7; IR (ATR): 3462, 2100, 1597, 1352, 1173, 1096, 982 cm−1; HRMS (EI) m/z: [M]+ calcd for C10H13N3O4S 271.0627, found 271.0622; HPLC analysis: Chiralpak AY-H, hexane/EtOH = 6/1, flow rate 1.0 mL/min, wavelength 254 nm, tR 28.9 min (minor) and 31.3 min (major).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27249025/s1, Figures S1–S10: Copies of 1H and 13C{1H} NMR spectra of compounds 2–12; Figures S11–S15: Chiral HPLC chromatogram of compounds 2, 5, 6, 7, and 8′.
Author Contributions
Conceptualization, O.O.; methodology, O.O.; investigation, K.Y., K.M., M.U. and Y.T.; writing—original draft preparation, K.Y.; writing—review and editing, O.O., M.K. and K.Y.; visualization, K.Y., K.M., M.U. and Y.T.; supervision, O.O.; project administration, K.Y. and M.K.; funding acquisition, O.O., M.K. and K.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by JSPS Grant-in-Aid for Scientific Research (22K06528, 22K15255, and 19K05459) and a grant from the Japan Soap and Detergent Association.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material.
Acknowledgments
The spectral data were collected with the research equipment shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for Supporting Introduction of the New Sharing System JPMXS0422500320).
Conflicts of Interest
The authors declare no competing financial interests.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Karmakar, B.; Halder, G. Progress and future of biodiesel synthesis: Advancements in oil extraction and conversion technologies. Energy Convers. Manag. 2019, 182, 307–339. [Google Scholar] [CrossRef]
- Pasha, M.K.; Dai, L.; Liu, D.; Guo, M.; Du, W. An overview to process design, simulation and sustainability evaluation of biodiesel production. Biotechnol. Biofuels 2021, 14, 129. [Google Scholar] [CrossRef] [PubMed]
- Quispe, C.A.G.; Coronado, C.J.R.; Carvalho, J.A. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; della Pina, C. From Glycerol to Value-Added Products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Beltramini, J.N.; Fan, Y.-X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Checa, M.; Nogales-Delgado, S.; Montes, V.; Encinar, J.M. Recent Advances in Glycerol Catalytic Valorization: A Review. Catalysts 2020, 10, 1279. [Google Scholar] [CrossRef]
- Furuta, T.; Sakai, M.; Hayashi, H.; Asakawa, T.; Kataoka, F.; Fujii, S.; Suzuki, T.; Suzuki, Y.; Tanaka, K.; Fishkin, N.; et al. Design and synthesis of artificial phospholipid for selective cleavage of integral membrane protein. Chem. Commun. 2005, 4575–4577. [Google Scholar] [CrossRef] [PubMed]
- Andresen, T.L.; Jensen, S.S.; Madsen, R.; Jørgensen, K. Synthesis and Biological Activity of Anticancer Ether Lipids That Are Specifically Released by Phospholipase A2 in Tumor Tissue. J. Med. Chem. 2005, 48, 7305–7314. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, L.; Provencal, D.P.; Miller, T.A.; O’Bryan, C.; Langston, M.; Shen, M.; Bailey, D.; Sha, D.; Palmer, T.; et al. Process Research and Kilogram Synthesis of an Investigational, Potent MEK Inhibitor. Org. Process Res. Dev. 2012, 16, 1652–1659. [Google Scholar] [CrossRef]
- Tangherlini, G.; Torregrossa, T.; Agoglitta, O.; Köhler, J.; Melesina, J.; Sippl, W.; Holl, R. Synthesis and biological evaluation of enantiomerically pure glyceric acid derivatives as LpxC inhibitors. Bioorg. Med. Chem. 2016, 24, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pasunooti, K.K.; Peng, H.; Li, R.-J.; Shi, W.Q.; Liu, W.; Cheng, Z.; Head, S.A.; Liu, J.O. Design and Synthesis of Tetrazole- and Pyridine-Containing Itraconazole Analogs as Potent Angiogenesis Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Tse, B. Total Synthesis of (−)-Galbonolide B and the Determination of Its Absolute Stereochemistry. J. Am. Chem. Soc. 1996, 118, 7094–7100. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Shiina, I.; Iwadare, H.; Saitoh, M.; Nishimura, T.; Ohkawa, N.; Sakoh, H.; Nishimura, K.; Tani, Y.; Hasegawa, M.; et al. Asymmetric Total Synthesis of Taxol®. Chem. Eur. J. 1999, 5, 121–161. [Google Scholar] [CrossRef]
- Byun, H.-S.; Sadlofsky, J.A.; Bittman, R. Enantioselective Synthesis of 3-Deoxy-(R)-sphingomyelin from (S)-1-(4’-Methoxyphenyl)glycerol. J. Org. Chem. 1998, 63, 2560–2563. [Google Scholar] [CrossRef]
- Reymond, S.; Cossy, J. Synthesis of migrastatin and its macrolide core. Tetrahedron 2007, 63, 5918–5929. [Google Scholar] [CrossRef]
- Yoshida, M.; Saito, K.; Kato, H.; Tsukamoto, S.; Doi, T. Total Synthesis and Biological Evaluation of Siladenoserinol A and its Analogues. Angew. Chem. Int. Ed. 2018, 57, 5147–5150. [Google Scholar] [CrossRef] [PubMed]
- Sigurjónsson, S.; Lúthersson, E.; Gudmundsson, H.G.; Haraldsdóttir, H.; Kristinsdóttir, L.; Haraldsson, G.G. Asymmetric Synthesis of Methoxylated Ether Lipids: A Glyceryl Glycidyl Ether Key Building Block Design, Preparation, and Synthetic Application. J. Org. Chem. 2022, 87, 12306–12314. [Google Scholar] [CrossRef]
- Lok, C.M.; Ward, J.P.; van Dorp, D.A. The synthesis of chiral glycerides starting from D- and L-serine. Chem. Phys. Lipids 1976, 16, 115–122. [Google Scholar]
- De Wilde, H.; De Clercq, P.; Vandewalle, M.; Röper, H. L-(S)-erythrulose a novel precursor to L-2,3-O-isopropylidene-C3 chirons. Tetrahedron Lett. 1987, 28, 4757–4758. [Google Scholar]
- Mikkilineni, A.B.; Kumar, P.; Abushanab, E. The Chemistry of L-Ascorbic and D-Isoascorbic Acids. 2. R and S Glyceraldehydes from a Common Intermediate. J. Org. Chem. 1988, 53, 6005–6009. [Google Scholar]
- Schmid, C.R.; Bryant, J.D.; Dowlatzedah, M.; Phillips, J.L.; Prather, D.E.; Schantz, R.D.; Sear, N.L.; Vianco, C.S. Synthesis of 2,3-O-Isopropylidene- D-Glyceraldehyde in High Chemical and Optical Purity: Observations on the Development of a Practical Bulk Process. J. Org. Chem. 1991, 56, 4056–4058. [Google Scholar]
- Doboszewski, B.; Herdewijn, P. Simple approach to 1-O-protected (R)- and (S)-glycerols from L- and D-arabinose for glycerol nucleic acids (GNA) monomers research. Tetrahedron Lett. 2011, 52, 3853–3855. [Google Scholar] [CrossRef]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Update 1 of: Enantioselective Enzymatic Desymmetrizations in Organic Synthesis. Chem. Rev. 2011, 111, PR110–PR180. [Google Scholar] [CrossRef] [PubMed]
- Chenault, H.K.; Chafin, L.F.; Liehr, S. Kinetic Chiral Resolutions of 1,2-Diols and Desymmetrization of Glycerol Catalyzed by Glycerol Kinase. J. Org. Chem. 1998, 63, 4039–4045. [Google Scholar] [CrossRef]
- Batovska, D.I.; Tsubota, S.; Kato, Y.; Asano, Y.; Ubukata, M. Lipase-mediated desymmetrization of glycerol with aromatic and aliphatic anhydrides. Tetrahedron Asymmetry 2004, 15, 3551–3559. [Google Scholar] [CrossRef]
- Caytan, E.; Cherghaoui, Y.; Barril, C.; Jouitteau, C.; Rabiller, C.; Remaud, G.S. Strategy for specific isotope ratio determination by quantitative NMR on symmetrical molecules: Application to glycerol. Tetrahedron Asymmetry 2006, 17, 1622–1624. [Google Scholar] [CrossRef]
- Franke, D.; Machajewski, T.; Hsu, C.-C.; Wong, C.-H. One-Pot Synthesis of L-Fructose Using Coupled Multienzyme Systems Based on Rhamnulose-1-phosphate Aldolase. J. Org. Chem. 2003, 68, 6828–6831. [Google Scholar] [CrossRef]
- Klibanov, A.M.; Alberti, B.N.; Marletta, M.A. Stereospecific Oxidation of Aliphatic Alcohols Catalyzed by Galactose Oxidase. Biochem. Biophys. Res. Commun. 1982, 108, 804–808. [Google Scholar] [CrossRef]
- Enríquez-García, Á.; Kündig, E.P. Desymmetrisation of meso-diols mediated by non-enzymatic acyl transfer catalysts. Chem. Soc. Rev. 2012, 41, 7803–7831. [Google Scholar] [CrossRef]
- Nájera, C.; Foubelo, F.; Sansano, J.M.; Yus, M. Enantioselective desymmetrization reactions in asymmetric catalysis. Tetrahedron 2022, 106–107, 132629. [Google Scholar] [CrossRef]
- Mizuta, S.; Sadamori, M.; Fujimoto, T.; Yamamoto, I. Asymmetric Desymmetrization of meso-1,2-Diols by Phosphinite Derivatives of Cinchona Alkaloids. Angew. Chem. Int. Ed. 2003, 42, 3383–3385. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rodrigo, J.; Hoveyda, A.H.; Snapper, M.L. Enantioselective silyl protection of alcohols catalysed by an amino-acid-based small molecule. Nature 2006, 443, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mitra, A.W.; Hoveyda, A.H.; Snapper, M.L. Kinetic Resolution of 1,2-Diols through Highly Site- and Enantioselective Catalytic Silylation. Angew. Chem. Int. Ed. 2007, 46, 8471–8474. [Google Scholar] [CrossRef] [PubMed]
- Demizu, Y.; Matsumoto, K.; Onomura, O.; Matsumura, Y. Copper complex catalyzed asymmetric monosulfonylation of meso-vic-diols. Tetrahedron Lett. 2007, 48, 7605–7609. [Google Scholar] [CrossRef]
- Sun, X.; Worthy, A.D.; Tan, K.L. Scaffolding Catalysts: Highly Enantioselective Desymmetrization Reactions. Angew. Chem. Int. Ed. 2011, 50, 8167–8171. [Google Scholar] [CrossRef]
- Hamaguchi, N.; Kuriyama, M.; Onomura, O. Chiral copper-catalyzed asymmetric monoarylation of vicinal diols with diaryliodonium salts. Tetrahedron Asymmetry 2016, 27, 177–181. [Google Scholar] [CrossRef]
- Li, R.-Z.; Tang, H.; Yang, K.R.; Wan, L.-Q.; Zhang, X.; Liu, J.; Fu, Z.; Niu, D. Enantioselective Propargylation of Polyols and Desymmetrization of meso 1,2-Diols by Copper/Borinic Acid Dual Catalysis. Angew. Chem. Int. Ed. 2017, 56, 7213–7217. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Michimuko, C.; Yamaguchi, K.; Nakajima, M.; Sugiura, M. Selective Monoacylation of Diols and Asymmetric Desymmetrization of Dialkyl meso-Tartrates Using 2-Pyridyl Esters as Acylating Agents and Metal Carboxylates as Catalysts. J. Org. Chem. 2019, 84, 9313–9321. [Google Scholar] [CrossRef]
- Trost, B.M.; Mino, T. Desymmetrization of Meso 1,3- and 1,4-Diols with a Dinuclear Zinc Asymmetric Catalyst. J. Am. Chem. Soc. 2003, 125, 2410–2411. [Google Scholar] [CrossRef]
- Honjo, T.; Nakao, M.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei, Y.; Nagao, Y. Nonenzymatic Enantioselective Monoacetylation of Prochiral 2-Protectedamino-2-Alkyl-1,3-Propanediols Utilizing a Chiral Sulfonamide-Zn Complex Catalyst. Org. Lett. 2007, 9, 509–512. [Google Scholar] [CrossRef]
- Lee, J.Y.; You, Y.S.; Kang, S.H. Asymmetric Synthesis of All-Carbon Quaternary Stereocenters via Desymmetrization of 2,2-Disubstituted 1,3-Propanediols. J. Am. Chem. Soc. 2011, 133, 1772–1774. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Tan, C.K.; Chen, F.; Yeung, Y.-Y. Catalytic Asymmetric Bromoetherification and Desymmetrization of Olefinic 1,3-Diols with C2-Symmetric Sulfides. J. Am. Chem. Soc. 2014, 136, 5627–5630. [Google Scholar] [CrossRef] [PubMed]
- Zi, W.; Toste, F.D. Gold(I)-Catalyzed Enantioselective Desymmetrization of 1,3-Diols through Intramolecular Hydroalkoxylation of Allenes. Angew. Chem. Int. Ed. 2015, 54, 14447–14451. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, J. Enantioselective Medium-Ring Lactone Synthesis through an NHC-Catalyzed Intramolecular Desymmetrization of Prochiral 1,3-Diols. ACS Catal. 2017, 7, 7647–7652. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tsuda, Y.; Kuriyama, M.; Demizu, Y.; Onomura, O. Copper-Catalyzed Enantioselective Synthesis of Oxazolines from Aminotriols via Asymmetric Desymmetrization. Chem. Asian. J. 2020, 15, 840–844. [Google Scholar] [CrossRef]
- Mandai, H.; Hironaka, T.; Mitsudo, K.; Suga, S. Acylative Desymmetrization of Cyclic meso-1,3-Diols by Chiral DMAP Derivatives. Chem. Lett. 2021, 50, 471–474. [Google Scholar] [CrossRef]
- Estrada, C.D.; Ang, H.T.; Vetter, K.-M.; Ponich, A.A.; Hall, D.G. Enantioselective Desymmetrization of 2-Aryl-1,3-Propanediols by Direct O-Alkylation with a Rationally Designed Chiral Hemiboronic Acid Catalyst That Mitigates Substrate Conformational Poisoning. J. Am. Chem. Soc. 2021, 143, 4162–4167. [Google Scholar] [CrossRef]
- Jung, B.; Hong, M.S.; Kang, S.H. Enantioselective Synthesis of Tertiary Alcohols by the Desymmetrizing Benzoylation of 2-Substituted Glycerols. Angew. Chem. Int. Ed. 2007, 46, 2616–2618. [Google Scholar] [CrossRef]
- Jung, B.; Kang, S.H. Chiral imine copper chloride-catalyzed enantioselective desymmetrization of 2-substituted 1,2,3-propanetriols. Proc. Natl. Acad. Sci. USA 2007, 104, 1471–1475. [Google Scholar] [CrossRef]
- You, Z.; Hoveyda, A.H.; Snapper, M.L. Catalytic Enantioselective Silylation of Acyclic and Cyclic Triols: Application to Total Syntheses of Cleroindicins D, F, and C. Angew. Chem. Int. Ed. 2009, 48, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Manville, N.; Alite, H.; Haeffner, F.; Hoveyda, A.H.; Snapper, M.L. Enantioselective silyl protection of alcohols promoted by a combination of chiral and achiral Lewis basic catalysts. Nat. Chem. 2013, 5, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Suganomata, Y.; Inoue, T.; Kuriyama, M.; Demizu, Y.; Onomura, O. Copper-Catalyzed Asymmetric Oxidative Desymmetrization of 2-Substituted 1,2,3-Triols. J. Org. Chem. 2022, 87, 6479–6491. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, J.; Asami, M.; Mukaiyama, T. An asymmetric synthesis of glycerol derivatives by the enantioselective acylation of prochiral glycerol. Chem. Lett. 1984, 13, 949–952. [Google Scholar] [CrossRef]
- Lewis, C.A.; Sculimbrene, B.R.; Xu, Y.; Miller, S.J. Desymmetrization of Glycerol Derivatives with Peptide-Based Acylation Catalysts. Org. Lett. 2005, 7, 3021–3023. [Google Scholar] [CrossRef]
- Trost, B.M.; Malhotra, S.; Mino, T.; Rajapaksa, N.S. Dinuclear Zinc-Catalyzed Asymmetric Desymmetrization of Acyclic 2-Substituted-1,3-Propanediols: A Powerful Entry into Chiral Building Blocks. Chem. Eur. J. 2008, 14, 7648–7657. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, A.; Umemura, S.; Ishihara, K. Desymmetrization of meso-Glycerol Derivatives Induced by L-Histidine-Derived Acylation Catalysts. Adv. Synth. Catal. 2011, 353, 1938–1942. [Google Scholar] [CrossRef]
- Giustra, Z.X.; Tan, K.L. The efficient desymmetrization of glycerol using scaffolding catalysis. Chem. Commun. 2013, 49, 4370–4372. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Tian, Y. Preparation of chiral glycerol sulfonate. Chinese Patent CN114394919A, 26 April 2022. [Google Scholar]
- Trost, B.M.; Older, C.M. A Convenient Synthetic Route to [CpRu(CH3CN)3]PF6. Organometallics 2002, 21, 2544–2546. [Google Scholar] [CrossRef]
- Hu, P.; Kan, J.; Su, W.; Hong, M. Pd(O2CCF3)2/Benzoquinone: A Versatile Catalyst System for the Decarboxylative Olefination of Arene Carboxylic Acids. Org. Lett. 2009, 11, 2341–2344. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, S.; Su, W.; Hong, M. Pd-Catalyzed Decarboxylative Heck Coupling with Dioxygen as the Terminal Oxidant. Org. Lett. 2010, 12, 4992–4995. [Google Scholar] [CrossRef] [PubMed]
- Onomura, O.; Takemoto, Y.; Miyamoto, K.; Ito, M. Method for preparing optically active 1,2,3-triol monoesters in the presence of bisoxazoline ligands and copper compounds. World Intellectual Property Organization WO2015/072290A1, 21 May 2015. [Google Scholar]
- Yokose, U.; Ishikawa, J.; Morokuma, Y.; Naoe, A.; Inoue, Y.; Yasuda, Y.; Tsujimura, H.; Fujimura, T.; Murase, T.; Hatamochi, A. The ceramide [NP]/[NS] ratio in the stratum corneum is a potential marker for skin properties and epidermal differentiation. BMC Dermatol. 2020, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Fukasawa, J.; Iwai, H.; Sugai, I.; Yamashita, O.; Kawamata, A. Multilamellar Emulsion of Stratum Corneum Lipid –Formation Mechanism and its Skin Care Effects. J. Soc. Cosmet. Chem. Japan 1993, 27, 193–205. [Google Scholar] [CrossRef]
- Ishida, K. Development and Properties of the Optically Active Ceramides. Oleoscience 2004, 4, 105–116. [Google Scholar] [CrossRef][Green Version]
- Maki, T.; Ushijima, N.; Matsumura, Y.; Onomura, O. Catalytic monoalkylation of 1,2-diols. Tetrahedron Lett. 2009, 50, 1466–1468. [Google Scholar] [CrossRef]
- Boldwin, J.J.; Raab, A.W.; Mensler, K.; Arison, B.H.; McClure, D.E. Synthesis of (R)- and (S)-Epichlorohydrin. J. Org. Chem. 1978, 43, 4876–4878. [Google Scholar] [CrossRef]
- Tanabe, G.; Sakano, M.; Minematsu, T.; Matusda, H.; Yoshikawa, M.; Muraoka, O. Synthesis and elucidation of absolute stereochemistry of salaprinol, another thiosugar sulfonium sulfate from the ayurvedic traditional medicine Salacia prinoides. Tetrahedron 2008, 64, 10080–10086. [Google Scholar] [CrossRef]
- Kurimura, M.; Takemoto, M.; Achiwa, K. Synthesis of Optically Active Lipopeptide Analogs from Outer membrane of Escherichia coli. Chem. Pharm. Bull. 1991, 39, 2590–2596. [Google Scholar] [CrossRef]
- Tuin, A.W.; Palachanis, D.K.; Buizert, A.; Grotenbreg, G.M.; Spalburg, E.; de Neeling, A.J.; Mars-Groenendijk, R.H.; Noort, D.; van der Marel, G.A.; Overkleeft, H.S.; et al. Synthesis and Biological Evaluation of Novel Gramicidin S Analogues. Eur. J. Org. Chem. 2009, 4231–4241. [Google Scholar] [CrossRef]
- Paterson, I.; Delgado, O.; Florence, G.J.; Lyothier, I.; O’Brien, M.; Scott, J.P.; Sereinig, N. A Second-Generation Total Synthesis of (+)-Discodermolide: The Development of a Practical Route Using Solely Substrate-Based Stereocontrol. J. Org. Chem. 2005, 70, 150–160. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).