
Unprecedented Epimerization of an Azithromycin Analogue: Synthesis, Structure and Biological Activity of 2′-Dehydroxy-5″-Epi-Azithromycin

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
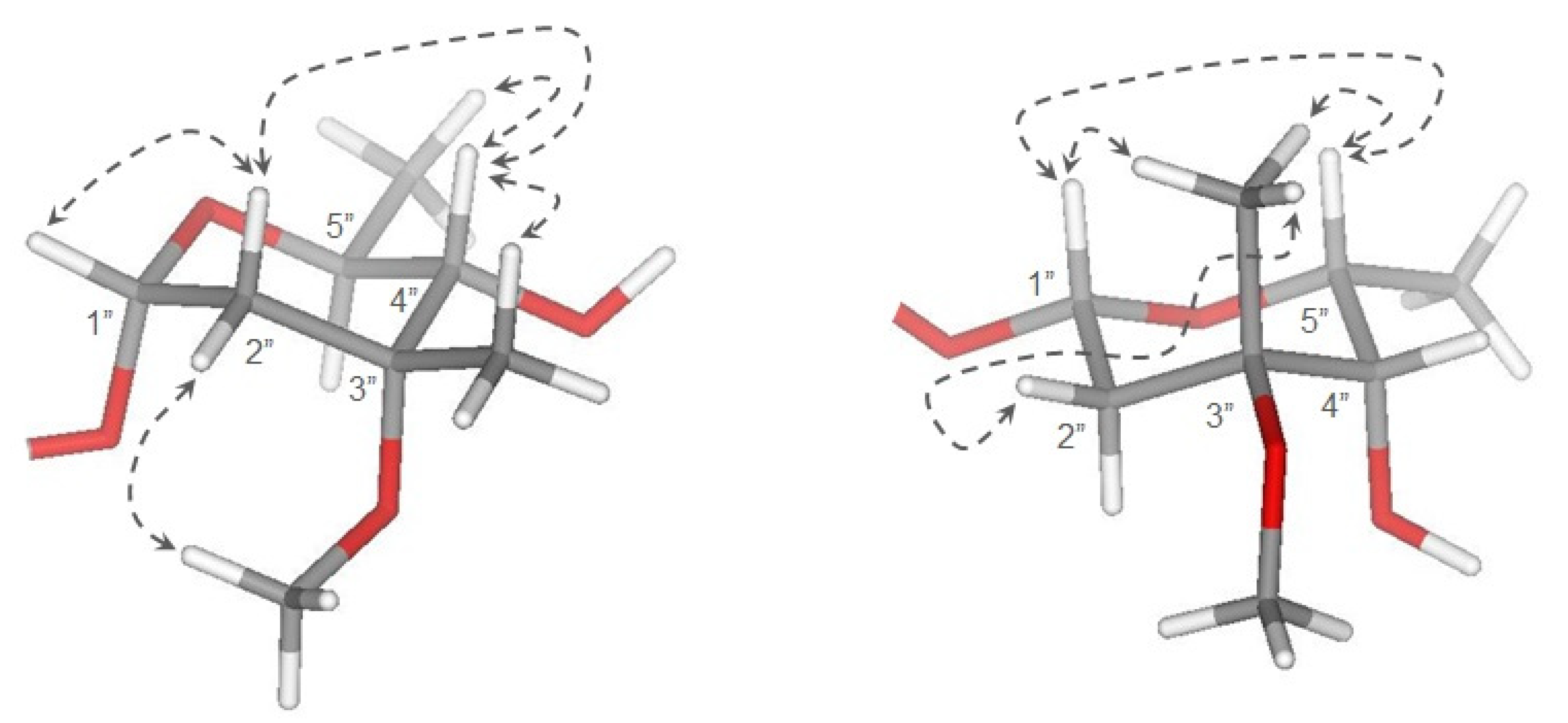
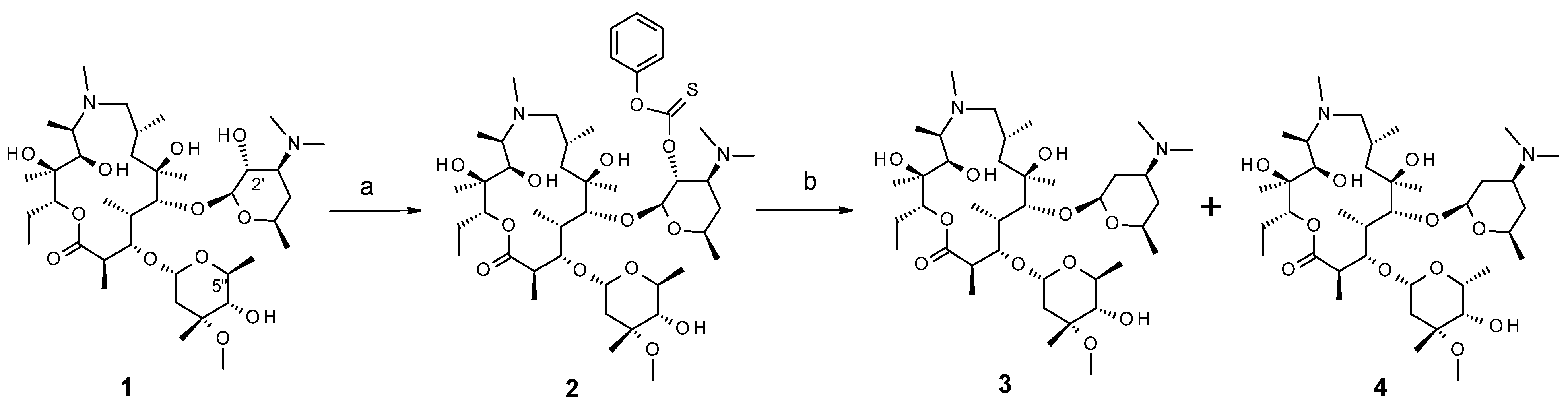
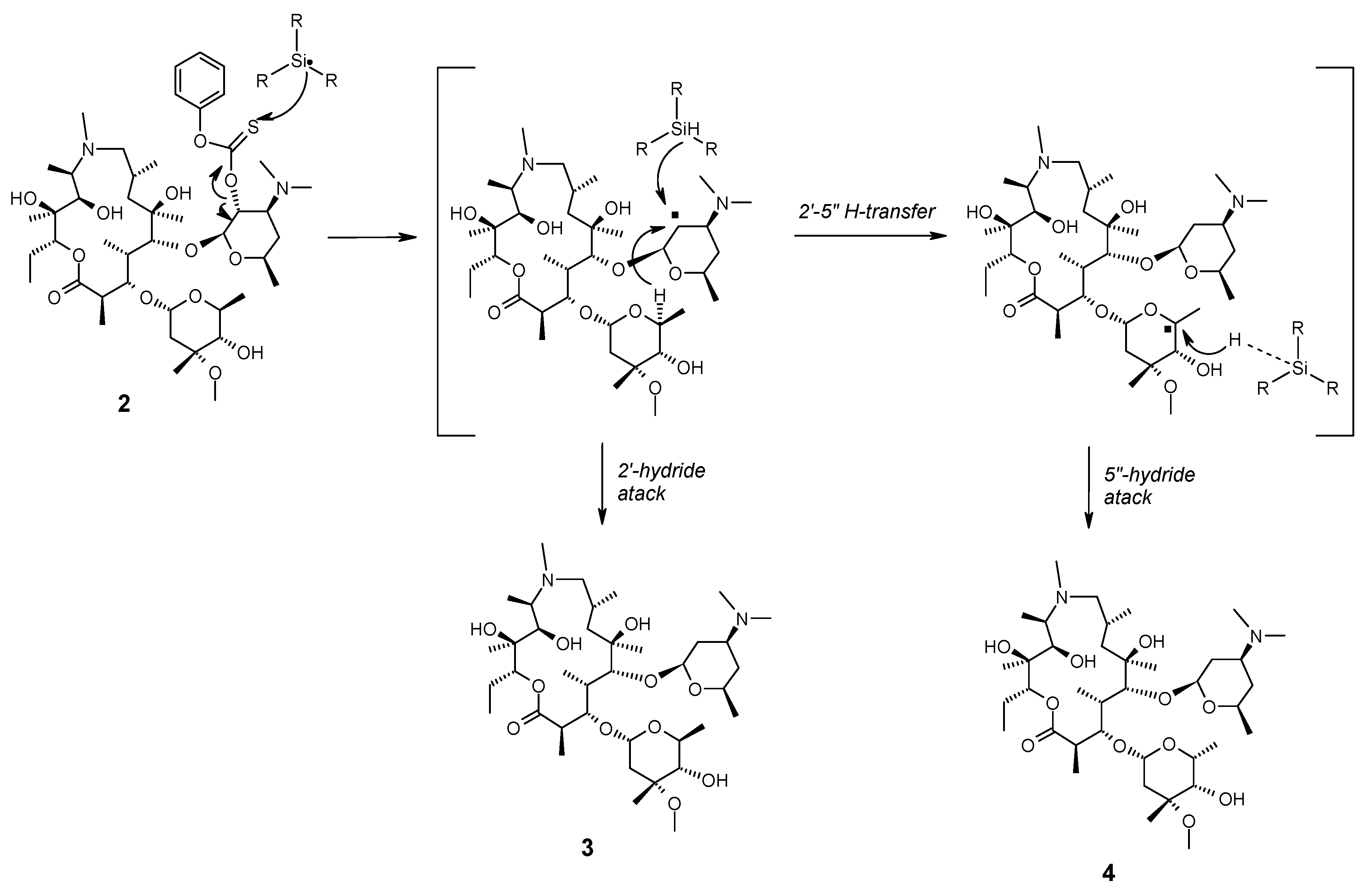
2.1. Chemistry

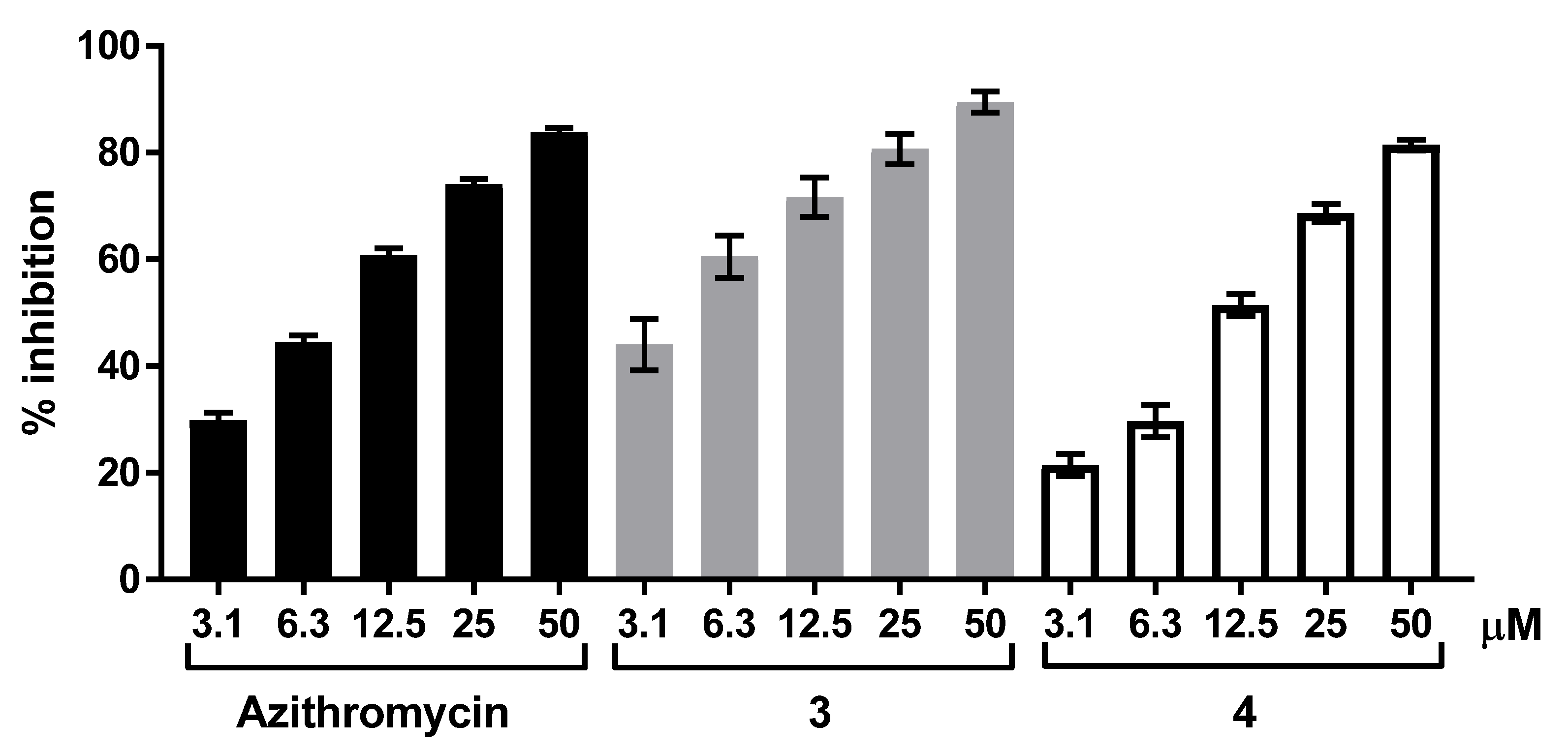
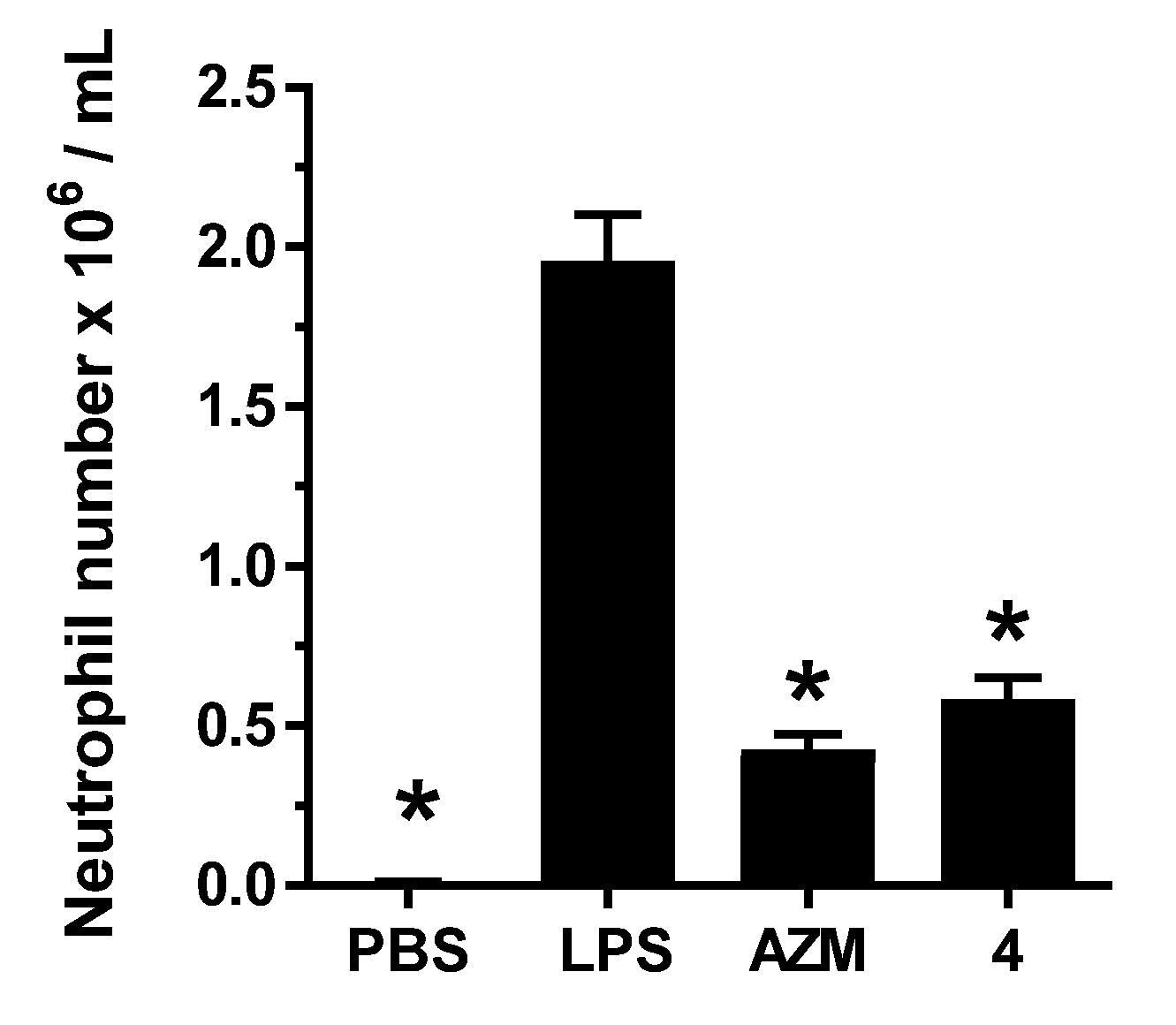
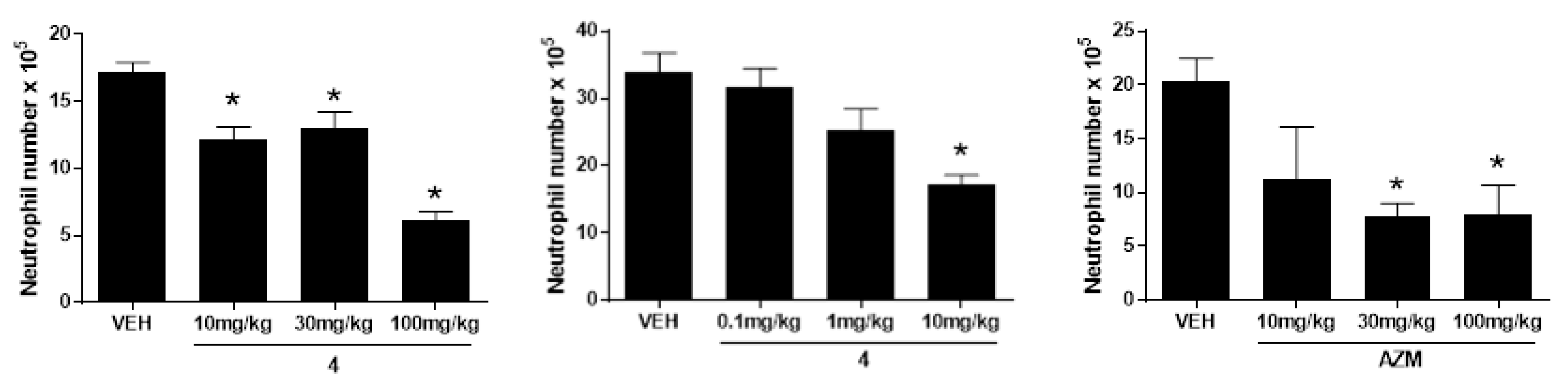
2.2. Biology
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. Antimicrobial Activity
3.2.2. LPS-Induced IL-6 Production by Murine Splenocytes
3.2.3. LPS-Induced Pulmonary Neutrophilia
3.2.4. Cigarette-Smoke-Induced Pulmonary Inflammation
3.2.5. Statistical Analysis
3.2.6. In Vitro Cytochrome Inhibition
3.2.7. Pharmacokinetics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parnham, M.J.; Erakovic, H.V.; Giamarellos-Bourboulis, E.J.; Perletti, G.; Verleden, G.M.; Vos, R. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol. Ther. 2014, 143, 225–245. [Google Scholar] [CrossRef]
- Cameron, E.J.; McSharry, C.; Chaudhuri, R.; Farrow, S.; Thomson, N.C. Long-term macrolide treatment of chronic inflammatory airway diseases: Risks, benefits and future developments. Clin. Exp. Allergy 2012, 42, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. Macrolides: From in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur. J. Clin. Pharmacol. 2012, 68, 479–503. [Google Scholar] [CrossRef] [PubMed]
- Bosnar, M.; Kragol, G.; Koštrun, S.; Vujasinović, I.; Bošnjak, B.; Mihaljević, V.B.; Ištuk, Z.M.; Kapić, S.; Hrvačić, B.; Brajša, K.; et al. N′-Substituted-2′-O,3′-N-carbonimidoyl bridged macrolides: Novel anti-inflammatory macrolides without antimicrobial activity. J. Med. Chem. 2012, 55, 6111–6123. [Google Scholar] [CrossRef]
- Bauer, J.; Vine, M.; Ćorić, I.; Bosnar, M.; Pašalić, I.; Turkalj, G.; Lazarevski, G.; Čulić, O.; Kragol, G. Impact of stereochemistry on the biological activity of novel oleandomycin derivatives. Bioorg. Med. Chem. 2012, 20, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Distrutti, E.; Renga, B.; Cipriani, S.; Palladino, G.; Booth, C.; Tudor, G.; Guse, J.-H.; Hahn, U.; Burnet, M.; et al. Development of non-antibiotic macrolide that corrects inflammation-driven immune dysfunction in models of inflammatory bowel diseases and arthritis. Eur. J. Pharmacol. 2011, 665, 29–39. [Google Scholar] [CrossRef]
- Sugawara, A.; Shima, H.; Sueki, A.; Hirose, T.; Matsui, H.; Nakano, H.; Hanaki, H.; Akagawa, K.S.; Ōmura, S.; Sunazuka, T. Non-antibiotic 12-membered macrolides: Design, synthesis and biological evaluation in a cigarette-smoking model. J. Antibiot. 2016, 69, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Menninger, J.R.; Otto, D.P. Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissociation of peptidyl-tRNA from ribosomes. Antimicrob. Agents Chemother. 1982, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, S.M.; Kofoed, C.; Vester, B. Inhibition of the ribosomal peptidyl transferase reaction by the mycarose moiety of the antibiotics carbomycin, spiramycin and tylosin. J. Mol. Biol. 2000, 304, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Champney, W.S.; Burdine, R. Macrolide antibiotics inhibit 50S ribosomal subunit assembly in Bacillus subtilis and Staphylo-coccus aureus. Antimicrob. Agents Chemother. 1995, 39, 2141–2144. [Google Scholar] [CrossRef] [Green Version]
- Usary, J.; Champney, W.S. Erythromycin inhibition of 50S ribosomal subunit formation in Escherichia coli cells. Mol. Microbiol. 2001, 40, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef]
- Mankin, A.S. Macrolide myths. Curr. Opin. Microbiol. 2008, 11, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Barber, J.; Čikoš, A.; Arsić, B.; Plavec, J.; Lazarevski, G.; Tepeš, P.; Košutić-Hulita, N. Free and bound state structures of 6-O-methyl homoerythromycins and epitope mapping of their interactions with ribosomes. Bioorg. Med. Chem. 2009, 17, 5857–5867. [Google Scholar] [CrossRef]
- Čikoš, A. Conformation and Binding Epitopes Determination in Macrolide Derivatives Interacting with 70S Escherichia coli Bacterial Ribosome by NMR Spectroscopy. Ph.D. Thesis, University of Zagreb, Zagreb, Croatia, 2011. [Google Scholar]
- Hansen, J.L.; Ippolito, J.A.; Ban, N.; Nissen, P.; Moore, P.B.; Steitz, T.A. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell 2002, 10, 117–128. [Google Scholar] [CrossRef]
- Tu, D.; Blaha, G.; Moore, P.B.; Steitz, T.A. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005, 121, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [Green Version]
- Barton, D.H.; McCombie, S.W.J. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc. Perkin Trans. 1975, 1, 1574–1585. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C. Progress of the barton-McCombie methodology: From tin hydrides to silanes. Res. Chem. Intermed. 1993, 19, 755–775. [Google Scholar] [CrossRef]
- McCombie, S.W.; Quiclet-Sire, B.; Zard, S.Z. Reflections on the mechanism of the Barton-McCombie deoxygenation and on its consequences. Tetrahedron 2018, 74, 4969–4979. [Google Scholar] [CrossRef]
- Novak, P.; Banić, T.Z.; Tepeš, P.; Lazarevski, G.; Plavec, J.; Turkalj, G. Conformational analysis of oleandomycin and its 8-methylene-9-oxime derivative by NMR and molecular modelling. Org. Biomol. Chem. 2005, 3, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Awan, A.; Brennan, R.J.; Regan, A.C.; Barber, J. The conformations of the macrolide antibiotics erythromycin A, azithromycin and clarithromycin in aqueous solution: A 1H NMR study. J. Chem. Soc. Perkin Trans. 2000, 2, 1645–1652. [Google Scholar] [CrossRef]
- Lazarevski, G.; Vinković, M.; Kobrehel, G.; Đokić, S.; Metelko, B.; Vikić-Topić, D. Conformational analysis of azithromycin by nuclear magnetic resonance spectroscopy and molecular modelling. Tetrahedron 1993, 49, 721–730. [Google Scholar] [CrossRef]
- Everett, J.R.; Tyler, J.W. The conformational analysis of erythromycin A. J. Chem. Soc. Perkin Trans. 1987, 2, 1659–1667. [Google Scholar] [CrossRef]
- Vester, B.; Douthwaite, S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 2001, 45, 1–12. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radical Quench/Eq. | Radical Initiator/Eq. | Concentration (Mol/L) | Temperature (°C) | Ratio of 3:4 |
|---|---|---|---|---|
| Bu3SnH/1.2 | AIBN/0.05 | 0.056 | 90 | 2.5:1 |
| Bu3SnH/5 | AIBN/0.05 | 0.11 | 90 | 1:0 |
| (TMS)3SiH/1.2 | AIBN/0.05 | 0.056 | 90 | 1:2 |
| (TMS)3SiH/1.2 | AIBN/0.05 | 0.023 | 90 | 1:3 |
| (TMS)3SiH/1.2 | ACCN/0.05 | 0.056 | 90 | 1:4 |
| (TMS)3SiH/1.1 | ACCN/0.5 | 0.035 | 115 | 1:12 |
| Atom | Cpd 3 | Cpd 4 | Atom | Cpd 3 | Cpd 4 |
|---|---|---|---|---|---|
| 2, 3 | 7.0 | 11.0 | 2, 2 CH3 | 7.7 | 7.5 |
| 3, 4 | 1.8 | 2.0 | 1″, 2″a | <1 | <1 |
| 4, 5 | 8.0 | 2.6 | 1″, 2″b | 4.9 | 11.1 |
| 7a, 8 | <1 | <1 | 4″, 5″ | 9.8 | <1 |
| 7b, 8 | 8.2 | 9.0 | 4, 4 CH3 | 7.7 | 7.7 |
| 8, 9a | 2.1 | 3.2 | 8, 8 CH3 | 7.2 | 7.2 |
| 8, 9b | 13.0 | 12.6 | 10, 10 CH3 | 7.2 | 7.2 |
| 10, 11 | <1 | 1.0 | 14, 15 | 7.7 | 7.6 |
| 13, 14a | 2.3 | 2.3 | 5′, 5′ CH3 | 6.2 | 6.4 |
| 13, 14b | 10.8 | 11.1 | 5″, 5″ CH3 | 6.4 | 6.9 |
| 1′, 2′a | 2.0 | 1.9 | |||
| 1′, 2′b | 9.5 | 9.8 | 7a, 7b | 15.9 | 17.2 |
| 2′a, 3′ | 4.0 | 4.0 | 9a, 9b | 13.0 | 12.6 |
| 2′b, 3′ | 13.1 | 13.1 | 14a, 14b | 14.6 | 14.6 |
| 3′, 4′a | 4.0 | 4.0 | 2′a, 2′b | 13.1 | 12.6 |
| 3′, 4′b | 12.8 | * | 4′a, 4′b | 13.0 | 12.8 |
| 4′a, 5′ | 1.8 | 1.8 | 2″a, 2″b | 16.4 | 12.6 |
| 4′b, 5′ | 11.3 | 11.3 |
| Compounds | S. aureus | S. penumoniae | S. pyogenes | M. catarrhalis | H. influenzae | E. coli |
|---|---|---|---|---|---|---|
| AZM | 0.5 | <0.125 | <0.125 | <0.125 | 2 | 2 |
| 3 | >64 | 32 | 64 | 16 | >64 | >64 |
| 4 | >64 | >64 | >64 | >64 | >64 | >64 |
| CL | Vss | t 1/2 | Oral F | |
|---|---|---|---|---|
| (mL/min/kg) | (L/kg) | (h) | (%) | |
| mouse | 4.1 ± 1.1 | 2 ± 0.8 | 10.5 ± 2.8 | 12.4 ± 4.2 |
| rat | 14.5 ± 8.5 | 11.7 ± 1.9 | 15.4 ± 6.1 | 27 ± 6 |
| dog | 5.5 ± 0.9 | 6.9 ± 2.0 | 20.5 ± 3.5 | 62 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kragol, G.; Steadman, V.A.; Marušić Ištuk, Z.; Čikoš, A.; Bosnar, M.; Jelić, D.; Ergović, G.; Trzun, M.; Bošnjak, B.; Bokulić, A.; et al. Unprecedented Epimerization of an Azithromycin Analogue: Synthesis, Structure and Biological Activity of 2′-Dehydroxy-5″-Epi-Azithromycin. Molecules 2022, 27, 1034. https://doi.org/10.3390/molecules27031034
Kragol G, Steadman VA, Marušić Ištuk Z, Čikoš A, Bosnar M, Jelić D, Ergović G, Trzun M, Bošnjak B, Bokulić A, et al. Unprecedented Epimerization of an Azithromycin Analogue: Synthesis, Structure and Biological Activity of 2′-Dehydroxy-5″-Epi-Azithromycin. Molecules. 2022; 27(3):1034. https://doi.org/10.3390/molecules27031034
Chicago/Turabian StyleKragol, Goran, Victoria A. Steadman, Zorica Marušić Ištuk, Ana Čikoš, Martina Bosnar, Dubravko Jelić, Gabrijela Ergović, Marija Trzun, Berislav Bošnjak, Ana Bokulić, and et al. 2022. "Unprecedented Epimerization of an Azithromycin Analogue: Synthesis, Structure and Biological Activity of 2′-Dehydroxy-5″-Epi-Azithromycin" Molecules 27, no. 3: 1034. https://doi.org/10.3390/molecules27031034
APA StyleKragol, G., Steadman, V. A., Marušić Ištuk, Z., Čikoš, A., Bosnar, M., Jelić, D., Ergović, G., Trzun, M., Bošnjak, B., Bokulić, A., Padovan, J., Glojnarić, I., & Eraković Haber, V. (2022). Unprecedented Epimerization of an Azithromycin Analogue: Synthesis, Structure and Biological Activity of 2′-Dehydroxy-5″-Epi-Azithromycin. Molecules, 27(3), 1034. https://doi.org/10.3390/molecules27031034