3.1. Chemistry
3.1.1. General Methods
All reagents and solvents were purchased from Sigma-Aldrich (Saint Louis, MO, USA), TCI (TCI Europe N.V., Zwijndrecht, Belgium), Combi-Blocks (San Diego, CA, USA) and used without further purification. All the reactions were monitored by thin-layer chromatography (TLC) or liquid chromatography–mass spectrometry (LCMS). TLC was carried out with Sigma-Aldrich silica gel (254 nm) plates (cat ref 99571-25EA) and TLC plates were revealed with UV light, KMnO4, p-anisaldehyde, or ninhydrin solutions. LCMS analysis was performed on an Agilent 1260 Infinity II UPLC machine (Agilent Technologies, Santa Clara, CA, USA) with a YMC-Triart C18 column (YMC CO., Kyoto, Japan). Flash chromatography purifications were performed on Biotage prepacked silica gel columns using Biotage Isolera instruments (Biotage, Upsala, Sweden). Reversed-phase preparative high-pressure liquid chromatography (HPLC) purification of final analogs was performed on a Waters Autopurification instrument (Waters Corporation, Milford, MA, USA) with MS- and UV-triggered collection operating at ambient temperature and at a flow rate of 16 mL/min. The identity of all compounds with reported biological activity was confirmed by NMR spectroscopy and low-resolution mass spectrometry (MS). The purity of all compounds with reported biological activity was ≥95% as determined by NMR and ultraperformance liquid chromatography (UPLC). Proton NMR spectra were recorded on a 300 or 400 MHz Bruker spectrometer (Bruker Corporation, Billerica, MA, USA) using TMS as internal standard, whereas carbon NMR spectra were recorded on a Bruker Avance 600 MHz instrument (Bruker Corporation, Billerica, MA, USA) (13C NMR, 150 MHz) using DMSO-d6 (39.5 ppm) as internal standard. Proton and carbon chemical shifts (δ) are reported in parts per million (ppm). Abbreviations used are: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Coupling constants are expressed in Hz. Low-resolution mass spectral data were obtained using a Waters H-Class UPLC (Waters Corporation, Milford, MA, USA) with a Waters Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 30 mm column (Waters Corporation, Milford, MA, USA), equipped with an Acquity UPLC PDA Detector (Waters Corporation, Milford, MA, USA), an Acquity UPLC ELS Detector (Waters Corporation, Milford, MA, USA)and an Acquity TQ Detector (ESI/ESCi) (Waters Corporation, Milford, MA, USA).
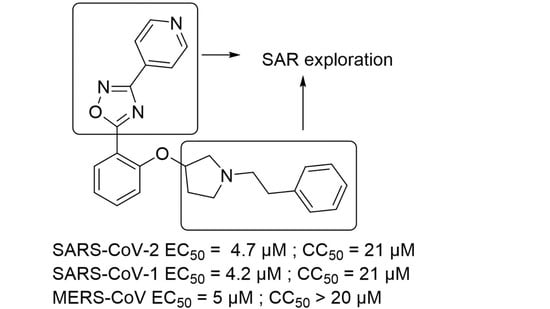
3.1.2. 2-(3-(Pyridin-4-yl)-1,2,4-oxadiazol-5-yl)phenol (4)
A solution of N′-hydroxypyridine-4-carboxamidine (0.500 g, 3.65 mmol, 1 eq) and salicylaldehyde (0.461 mL, 4.38 mmol, 1.2 eq) in EtOH (1.5 mL) was irradiated in a microwave oven at 160 °C for 1 h. After cooling to room temperature, the beige solid was filtered, washed with cold ethanol and dried under high vacuum to afford 0.228 g (26%) of the title compound. LC-MS (ESI, m/z): 240 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.69 (s, 1H), 8.84 (d, J = 6.1 Hz, 2H), 8.02–8.05 (m, 3H), 7.52–7.60 (m, 1H), 7.15 (d, J = 8.5 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H).
3.1.3. 5-(2-((1-Phenethylpyrrolidin-3-yl)oxy)phenyl)-3-(pyridin-4-yl)-1,2,4-oxadiazole (1)
A mixture of pyrrolidin-3-ol (0.100 g, 1.11 mmol, 1 eq), (2-bromoethyl) benzene (0.307 mL, 2.23 mmol, 2 eq) and K2CO3 (0.769 g, 5.57 mmol, 5 eq) in toluene (5 mL) was heated at 110 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was taken up with 1N HCl and extracted with CH2Cl2 twice. The aqueous phase was basified by the addition of K2CO3 and extracted with CH2Cl2. The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure to afford 0.130 g (61%) of 1-(2-phenylethyl)pyrrolidin-3-ol as a white solid. LC-MS (ESI, m/z): 192 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.12–7.33 (m, 5H), 4.67 (d, J = 4.6 Hz, 1H), 4.12–4.23 (m, 1H), 2.65–2.80 (m, 3H), 2.55–2.64 (m, 3H), 2.41–2.48 (m, 1H), 2.34 (m, 1H), 1.90–2.02 (m, 1H), 1.45–1.58 (m, 1H).
To a solution of
4 (0.050 g, 0.209 mmol, 1 eq), 1-(2-phenylethyl)pyrrolidin-3-ol (0.056 g, 0.293 mmol, 1.4 eq) and 1,1′-(azodicarbonyl)dipiperidine (0.075 g, 0.293 mmol, 1.4 eq) in THF (2.2 mL) was added
nBu
3P (0.079 mL, 0.293 mmol, 1.4 eq). The reaction mixture was stirred at room temperature for 2 d and was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (0–15%) in CH
2Cl
2. A second purification by flash chromatography on silica gel using a gradient of MeOH (0–7%) in CH
2Cl
2 followed by trituration with Et
2O furnished 0.045 g (51%) of the title compound as a white solid. LC-MS (ESI,
m/
z): 413 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.84 (d,
J = 4.8 Hz, 2H), 8.12 (d,
J = 7.3 Hz, 1H), 8.02 (d,
J = 5.7 Hz, 2H), 7.68 (t,
J = 7.8 Hz, 1H), 7.11–7.32 (m, 7H), 5.11 (m, 1H), 3.06 (m, 1H), 2.74–2.86 (m, 4H), 2.67 (m, 2H), 2.59 (m, 1H), 2.34 (m, 1H), 1.94 (m, 1H) (
Figure S1).
13C NMR (150 MHz, DMSO-
d6) δ 175.89, 166.29, 156.64, 150.99, 140.38, 135.10, 133.74, 131.63, 128.65, 128.27, 125.92, 121.08, 120.96, 114.81, 112.68, 77.72, 59.63, 57.32, 52.50, 34.53, 31.87 (
Figure S2).
3.1.4. 5-[2-[(3R)-1-(2-Phenylethyl)pyrrolidin-3-yl]oxyphenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (1a)
The title compound was prepared similarly to
1 using (3S)-1-(2-phenylethyl)pyrrolidin-3-ol instead of pyrrolidin-3-ol (
Figures S3 and S4).
3.1.5. 5-[2-[(3S)-1-(2-Phenylethyl)pyrrolidin-3-yl]oxyphenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (1b)
The title compound was prepared similarly to
1 using (3
R)-1-(2-phenylethyl)pyrrolidin-3-ol instead of pyrrolidin-3-ol (
Figure S5).
3.1.6. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (8)
A mixture of pyrrolidin-3-ol (0.100 g, 1.11 mmol, 1 eq), bromocyclopentane (0.244 mL, 2.23 mmol, 2 eq) and K2CO3 (0.769 g, 5.57 mmol, 5 eq) in toluene (5 mL) was heated at 110 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was taken up with 1N HCl and extracted with CH2Cl2 twice. The aqueous phase was basified by the addition of K2CO3 and extracted with CH2Cl2. The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure to afford 0.081 g (47%) of 1-cyclopentylpyrrolidin-3-ol as a white solid. LC-MS (ESI, m/z): 156 [M + H]+.
The title compound was prepared similarly to
1 using 1-cyclopentylpyrrolidin-3-ol instead of 1-(2-phenylethyl)pyrrolidin-3-ol. LC-MS (ESI,
m/
z): 377 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.84 (m, 2H), 8.12 (dd,
J = 1.5, 7.6 Hz, 1H), 8.02 (m, 2H), 7.68 (m, 1H), 7.26 (d,
J = 8.5 Hz, 1H), 7.19 (t,
J = 7.6 Hz, 1H), 5.07 (t,
J = 6.7 Hz, 1H), 3.02 (m, 1H), 2.65–2.77 (m, 2H), 2.43–2.48 (m, 2H), 2.33 (m, 1H), 1.92 (m, 1H), 1.73 (m, 2H), 1.33–1.67 (m, 6H) (
Figure S6).
3.1.7. 5-[2-[1-(Cyclopentylmethyl)pyrrolidin-3-yl]oxyphenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (9)
To a solution of 4 (0.165 g, 0.690 mmol, 1 eq) in THF (2.2 mL) were added DIAD (0.217 mL, 1.03 mmol, 1.5 eq), 1-Boc-3-hydroxypyrrolidine (0.200 g, 1.03 mmol, 1.5 eq) and PPh3 (0.271 g, 1.03 mmol, 1.5 eq). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with EtOAc and washed with water. The phases were separated. The organic phase was washed with 1N NaOH, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (20–60%) in heptane to afford 0.276 g (98%) of tert-butyl 3-[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]pyrrolidine-1-carboxylate as a white solid. LC-MS (ESI, m/z): 409 [M + H]+.
To a mixture of tert-butyl 3-[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]pyrrolidine-1-carboxylate (0.276 g, 0.676 mmol, 1 eq) in dioxane (2 mL) was added 4N HCl in dioxane (1.01 mL; 4.04 mmol, 6 eq). The reaction mixture was stirred at room temperature for 4 h and was concentrated under reduced pressure to afford quantitatively 3-(4-pyridyl)-5-(2-pyrrolidin-3-yloxyphenyl)-1,2,4-oxadiazole dihydrochloride as a white solid.
To a solution of 3-(4-pyridyl)-5-(2-pyrrolidin-3-yloxyphenyl)-1,2,4-oxadiazole dihydrochloride (0.100 g, 0.262 mmol, 1 eq) in DMF (1 mL) were added K
2CO
3 (0.073 g, 0.525 mmol, 2 eq) and iodomethylcyclopentane (0.053 mL, 0.393 mmol, 1.5 eq). The reaction mixture was heated at 80 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with CH
2Cl
2. The organic phase was dried over MgSO
4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (0–25%) in CH
2Cl
2. A second purification by flash chromatography on C18 using a gradient of CH
3CN (5–100%) in water furnished 0.043 g (40%) of the title compound as a beige solid. LC-MS (ESI,
m/
z): 391 [M + H]
+.
1H NMR (400 MHz, CD
3OD) δ 8.78 (m, 2H), 8.14 (m, 3H), 7.66 (m, 1H), 7.20 (m, 2H), 5.12 (m, 1H), 3.14 (m, 1H), 2.93 (m, 1H), 2.85 (m, 1H), 2.70 (m, 1H), 2.45–2.60 (m, 2H), 2.40 (m, 1H), 2.01–2.18 (m, 2H), 1.83 (m, 2H), 1.50–1.72 (m, 4H), 1.24 (m, 2H) (
Figure S7).
13C NMR (150 MHz, DMSO-
d6) δ 175.55, 166.30, 156.68, 150.97, 135.07, 133.76, 131.57, 121.07, 120.91, 114.83, 112.70, 77.80, 61.46, 59.96, 52.84, 38.47, 31.86, 30.80, 24.81 (
Figure S8).
3.1.8. 5-[2-[(1-Cyclopentylpyrrolidin-3-yl)methoxy]phenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (10)
To a solution of 4 (0.300 g, 1.25 mmol, 1 eq) in THF (4 mL) were added DIAD (0.394 mL, 1.88 mmol, 1.5 eq), tert-butyl 3-(hydroxymethyl)pyrrolidine-1-carboxylate (0.379 g, 1.88 mmol, 1.5 eq) and PPh3 (0.493 g, 1.88 mmol, 1.5 eq). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with EtOAc and washed with water. The phases were separated. The organic phase was washed with 1N NaOH, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (20–60%) in heptane to afford 0.529 g (99%) of tert-butyl 3-[[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]methyl]pyrrolidine-1-carboxylate as a white solid. LC-MS (ESI, m/z): 423 [M + H]+.
To a mixture of tert-butyl 3-[[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]methyl]pyrrolidine-1-carboxylate (0.529 g, 1.25 mmol, 1 eq) in dioxane (4 mL) was added 4N HCl in dioxane (1.01 mL; 4.04 mmol, 3.2 eq). The reaction mixture was stirred at room temperature overnight and was concentrated under reduced pressure to afford quantitatively 3-(4-pyridyl)-5-[2-(pyrrolidin-3-ylmethoxy)phenyl]-1,2,4-oxadiazole dihydrochloride as a white solid.
To a solution of 3-(4-pyridyl)-5-[2-(pyrrolidin-3-ylmethoxy)phenyl]-1,2,4-oxadiazole dihydrochloride (0.100 g, 0.253 mmol, 1 eq) in DMF (2 mL) were added K
2CO
3 (0.175 g, 1.26 mmol, 5 eq), NaI (0.019 g, 0.127 mmol, 0.5 eq) and bromocyclopentane (0.083 mL, 0.759 mmol, 3 eq). The reaction mixture was heated at 80 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with CH
2Cl
2. The organic phase was dried over MgSO
4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (0–25%) in CH
2Cl
2 to afford 0.026 g (26%) of the title compound as a beige solid. LC-MS (ESI,
m/
z): 391 [M + H]
+.
1H NMR (400 MHz, CD
3OD) δ 8.81 (d,
J = 5.8 Hz, 2H), 8.24 (m, 1H), 8.14 (m, 2H), 7.71 (m, 1H), 7.33 (d,
J = 8.5 Hz, 1H), 7.24 (t,
J = 7.6 Hz, 1H), 4.27–4.39 (m, 2H), 3.75 (m, 1H), 3.36–3.62 (m, 4H), 3.07 (m, 1H), 2.38 (m, 1H), 2.18 (m, 3H), 1.86 (m, 2H), 1.71 (m, 4H) (
Figure S9).
13C NMR (150 MHz, DMSO-
d6) δ 175.57, 166.49, 157.41, 151.05, 135.29, 133.70, 131.31, 121.41, 121.12, 114.07, 112.23, 69.70, 65.77, 54.61, 52.10, 36.15, 29.03, 26.02, 23.58 (
Figure S10).
3.1.9. 5-[2-[[1-(Cyclopentylmethyl)pyrrolidin-3-yl]methoxy]phenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (11)
The title compound was prepared similarly to
10 using iodomethylcyclopentane instead of bromocyclopentane. LC-MS (ESI,
m/
z): 405 [M + H]
+.
1H NMR (400 MHz, CD
3OD) δ 8.80 (m, 2H), 8.20 (dd,
J = 1.5, 7.5 Hz, 1H), 8.15 (m, 2H), 7.68 (m, 1H), 7.30 (d,
J = 8.5 Hz, 1H), 7.21 (t,
J = 7.5 Hz, 1H), 4.15–do4.32 (m, 2H), 3.12 (m, 1H), 2.85–3.04 (m, 3H), 2.80 (m, 2H), 2.08–2.30 (m, 2H), 1.81–2.00 (m, 3H), 1.52–1.73 (m, 4H), 1.17–1.39 (m, 3H) (
Figure S11).
13C NMR (150 MHz, DMSO-
d6) δ 175.71, 166.40, 157.68, 151.00, 135.22, 133.74, 131.30, 121.17, 121.08, 114.06, 112.24, 71.06, 60.66, 56.60, 53.53, 37.78, 36.39, 30.77, 26.45, 24.75 (
Figure S12).
3.1.10. 5-[2-[(1-Cyclopentyl-4-piperidyl)oxy]phenyl]-3-(4-pyridyl)-1,2,4-oxadiazole (12)
To a solution of 4 (0.165 g, 0.690 mmol, 1 eq) in THF (2.2 mL) were added DIAD (0.217 mL, 1.03 mmol, 1.5 eq), 1-Boc-4-hydroxypiperidine (0.213 g, 1.03 mmol, 1.5 eq) and PPh3 (0.271 g, 1.03 mmol, 1.5 eq). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with EtOAc and washed with water. The phases were separated. The organic phase was washed with 1N NaOH, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (20–60%) in heptane to afford 0.276 g (95%) of tert-butyl 4-[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]piperidine-1-carboxylate as a white solid. LC-MS (ESI, m/z): 423 [M + H]+.
To a mixture of tert-butyl 4-[[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]piperidine-1-carboxylate (0.271 g, 0.642 mmol, 1 eq) in dioxane (2 mL) was added 4N HCl in dioxane (0.962 mL; 3.85 mmol, 6 eq). The reaction mixture was stirred at room temperature overnight and was concentrated under reduced pressure to afford quantitatively 5-[2-(4-piperidyloxy)phenyl]-3-(4-pyridyl)-1,2,4-oxadiazole dihydrochloride as a white solid.
To a solution of 5-[2-(4-piperidyloxy)phenyl]-3-(4-pyridyl)-1,2,4-oxadiazole dihydrochloride (0.100 g, 0.253 mmol, 1 eq) in DMF (2 mL) were added K
2CO
3 (0.175 g, 1.26 mmol, 5 eq), NaI (0.019 g, 0.127 mmol, 0.5 eq) and bromocyclopentane (0.083 mL, 0.759 mmol, 3 eq). The reaction mixture was heated at 80 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with CH
2Cl
2. The organic phase was dried over MgSO
4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (1–25%) in CH
2Cl
2 to afford 0.056 g (57%) of the title compound as a beige solid. LC-MS (ESI,
m/
z): 391 [M + H]
+.
1H NMR (400 MHz, CD
3OD) δ 8.80 (m, 2H), 8.21 (dd,
J = 1.5, 7.9 Hz, 1H), 8.16 (m, 2H), 7.68 (m, 1H), 7.36 (d,
J = 8.2 Hz, 1H), 7.22 (t,
J = 7.9 Hz, 1H), 4.97 (m, 1H), 3.23 (m, 2H), 3.03–3.18 (m, 3H), 2.17 (m, 4H), 2.07 (m, 2H), 1.79 (m, 2H), 1.63 (m, 4H) (
Figure S13).
13C NMR (150 MHz, DMSO-
d6) δ 175.70, 166.48, 156.00, 151.00, 135.11, 133.78, 131.56, 121.29, 121.14, 115.45, 113.20, 71.21, 66.65, 47.47, 28.87, 23.66 (
Figure S14).
3.1.11. 5-(2-(2-Bromoethoxy)phenyl)-3-(pyridin-4-yl)-1,2,4-oxadiazole (7)
To a solution of 4 (3.0 g, 12.5 mmol, 1 eq) in DMF (30 mL) were added K2CO3 (3.5 g, 25.1 mmol, 2 eq) and 1,2-dibromoethane (3.24 g, 37.6 mmol, 3 eq). The reaction mixture was heated at 80 °C for 2 h. After cooling to room temperature, the reaction mixture was poured into ice/water. The resulting precipitate was filtered and dried under high vacuum to afford 2.0 g (46%) of the title compound as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 8.84 (d, J = 5.9 Hz, 2H), 8.12 (m, 1H), 8.03 (d, J = 5.8 Hz, 2H), 7.70 (m, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.23 (t, J = 7.5 Hz, 1H), 4.55 (t, J = 5.2 Hz, 2H), 3.88 (t, J = 5.2 Hz, 2H).
3.1.12. N-[2-[2-[3-(4-Pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]ethyl]cyclopentanamine (13)
To a solution of
7 (0.200 g, 0.578 mmol, 1 eq) in DMF (2 mL) were added K
2CO
3 (0.160 g, 1.16 mmol, 2 eq) and cyclopentanamine (0.074 g, 0.867 mmol, 1.5 eq). The reaction mixture was heated at 80 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with 10% MeOH in CH
2Cl
2. The organic phase was washed with brine, dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Xbridge C18 column,250 × 19 mm, 10µ; 20 to 65% ACN in 20 mM ammonium bicarbonate in 35 min) to afford 0.040 g (20%) of the title compound as an off-white solid. LC-MS (ESI,
m/
z): 351 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.84 (d,
J = 4.9 Hz, 2H), 8.10 (d,
J = 7.3 Hz, 1H), 8.00 (d,
J = 4.9 Hz, 2H), 7.69 (t,
J = 7.8 Hz, 1H), 7.33 (d,
J = 7.8 Hz, 1H), 7.19 (t,
J = 7.3 Hz, 1H), 4.23 (m, 2H), 3.09 (m, 1H), 2.94 (m, 2H), 1.70 (m, 2H), 1.58 (m, 2H), 1.44 (m, 2H), 1.28 (m, 2H) (NH not visible) (
Figure S15).
13C NMR (150 MHz, DMSO-
d6) δ 175.63, 166.45, 157.72, 150.95, 135.23, 133.73, 131.22, 121.09, 114.05, 112.21, 68.89, 58.94, 46.70, 32.80, 23.58 (
Figure S16).
3.1.13. N-Methyl-N-[2-[2-[3-(4-pyridyl)-1,2,4-oxadiazol-5-yl]phenoxy]ethyl]cyclopentanamine (14)
The title compound was prepared similarly to
13 using
N-methylcyclopentanamine instead of cyclopentanamine. LC-MS (ESI,
m/
z): 365 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.84 (d,
J = 5.9 Hz, 2H), 8.08 (d,
J = 7.3 Hz, 1H), 8.01 (d,
J = 5.9 Hz, 2H), 7.69 (t,
J = 7.8 Hz, 1H), 7.35 (d,
J = 7.8 Hz, 1H), 7.18 (t,
J = 7.3 Hz, 1H), 4.24 (m, 2H), 2.83 (m, 2H), 2.75 (m, 1H), 2.26 (s, 3H), 1.71 (m, 2H), 1.54 (m, 2H), 1.43 (m, 2H), 1.33 (m, 2H) (
Figure S17).
13C NMR (150 MHz, DMSO-
d6) δ 175.83, 166.36, 157.75, 150.98, 135.12, 133.73, 131.36, 121.08, 120.93, 113.94, 112.30, 67.65, 66.21, 53.98, 40.80, 29.92, 23.84 (
Figure S18).
3.1.14. tert-Butyl 3-(2-(methoxycarbonyl)phenoxy)pyrrolidine-1-carboxylate (17)
To a solution of DIAD (8.8 mL, 44.9 mmol, 1.2 eq) in THF (20 mL) cooled at 0 °C was added PPh3 (10.5 g, 41.1 mmol, 1.1 eq). The reaction mixture was stirred at 0 °C for 30 min. tert-Butyl 3-hydroxypyrrolidine-1-carboxylate (7.0 g, 37.4 mmol, 1 eq) was added and the reaction mixture was stirred at 0 °C for 15 min. Methyl 2-hydroxybenzoate (5.7 g, 37.4 mmol, 1 eq) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between water and EtOAc. The phases were separated. The aqueous phase was extracted twice with EtOAc. The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (0–50%) in hexane to afford 4.0 g (33%) of the title compound as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 1.6, 7.6 Hz, 1H), 7.42 (m, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 4.94 (m, 1H), 3.85 (s, 3H), 3.52–3.60 (m, 4H), 2.22 (m, 1H), 2.07 (m, 1H), 1.45 (s, 9H).
3.1.15. Methyl 2-(pyrrolidin-3-yloxy)benzoate hydrochloride (18)
To a solution of 17 (12.8 g, 39.7 mmol, 1 eq) in CH2Cl2 (130 mL) cooled at 0 °C was added 4N HCl in dioxane (30 mL, 120 mmol, 3 eq). The reaction mixture was stirred at room temperature overnight and was concentrated under reduced pressure to afford quantitatively the title compound as a brown oil. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (br. s., 1H), 9.30 (br. s., 1H), 7.70 (dd, J = 1.6, 7.6 Hz, 1H), 7.56 (t, J = 8.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 5.22 (m, 1H), 3.81 (s, 3H), 3.49 (m, 1H), 3.15–3.40 (m, 3H), 2.14 (m, 2H).
3.1.16. Methyl 2-((1-cyclopentylpyrrolidin-3-yl)oxy)benzoate (19)
To a solution of 18 (11.4 g, 44.2 mmol, 1 eq) in CH2Cl2 (120 mL) was added cyclopentanone (5.1 mL, 57.5 mmol, 1.3 eq) and the reaction mixture was stirred at room temperature for 3 h. NaBH(OAc)3 (14.1 g, 66.4 mmol, 1.5 eq) was added portion-wise and the reaction mixture was stirred at room temperature overnight. The reaction mixture was washed with water and the phases were separated. The aqueous phase was extracted with 10% MeOH in CH2Cl2 (3×). The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (0–10%) in CH2Cl2 to afford 9.9 g (77%) of the title compound as a brown oil. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 6.8 Hz, 1H), 7.43 (t, J = 7.2 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 5.00 (m, 1H), 3.85 (s, 3H), 3.74 (m, 1H), 3.29 (m, 1H), 3.10 (m, 1H), 2.92–3.02 (m, 2H), 2.34 (m, 1H), 2.15 (m, 1H), 1.93 (m, 3H), 1.76 (m, 3H), 1.55 (m, 2H).
3.1.17. Lithium 2-((1-cyclopentylpyrrolidin-3-yl)oxy)benzoate (20)
To a solution of 19 (1.0 g, 3.46 mmol, 1 eq) in THF (30 mL), MeOH (10 mL) and water (20 mL) was added LiOH (0.138 g, 3.28 mmol, 0.9 eq). The reaction mixture was heated at 60 °C for 6 h and was concentrated under reduced pressure. The residue was dissolved in water (30 mL) and was washed twice with CH2Cl2. The aqueous phase was lyophilized to afford 0.800 g (82%) of lithium 2-((1-cyclopentylpyrrolidin-3-yl)oxy)benzoate as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.15 (d, J = 7.2 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.75 (m, 2H), 4.75 (m, 1H), 2.80 (m, 1H), 2.57–2.66 (m, 2H), 2.41 (m, 2H), 2.13 (m, 1H), 1.70–1.85 (m, 3H), 1.58–1.64 (m, 2H), 1.30–1.55 (m, 4H).
3.1.18. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(3-pyridyl)-1,2,4-oxadiazole (23)
To a solution of
N-hydroxynicotinimidamide (0.150 g, 1.09 mmol, 1 eq) in DMSO (2 mL) were added
19 (0.474 g, 1.64 mmol, 1.5 eq) and NaOH (0.066 g, 1.64 mmol, 1.5 eq). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic phase was dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (10–20%) in EtOAc. A second purification by reversed-phase HPLC (Xterra RP18 column,250 × 19 mm, 10µ; 20 to 85% MeOH in 20 mM ammonium bicarbonate in 24 min) furnished 0.015 g (4%) of the title compound as an off-white solid. LC-MS (ESI,
m/
z): 377 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 9.26 (s, 1H), 8.81 (d,
J = 3.9 Hz, 1H), 8.44 (d,
J = 7.3 Hz, 1H), 8.12 (d,
J = 7.3 Hz, 1H), 7.67 (m, 2H), 7.09–7.38 (m, 2H), 5.07 (m, 1H), 3.01 (m, 1H), 2.70 (m, 2H), 2.33 (m, 2H), 1.89 (m, 1H), 1.73 (m, 2H), 1.60 (m, 2H), 1.31–1.54 (m, 5H) (
Figure S19).
3.1.19. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-pyrimidin-5-yl-1,2,4-oxadiazole (24)
A mixture of 20 (0.250 g, 0.889 mmol, 1 eq), HATU (0.406 g, 1.07 mmol, 1.2 eq) and DIPEA (0.372 mL, 2.14 mmol, 2.4 eq) in DMF (3 mL) was stirred at room temperature for 15 min. After the addition of
N-hydroxypyrimidine-5-carboximidamide (0.123 g, 0.890 mmol, 1 eq) [
28], the reaction mixture was stirred at room temperature for 6 h and was heated at 100 °C overnight. After cooling to room temperature, the reaction mixture was diluted with CH
2Cl
2 and was washed with sat. NaHCO
3. The phases were separated. The organic phase was washed with sat. NaHCO
3, dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (YMC-Actus C18 column,250 × 20 mm, 5µ; 40 to 90% ACN in 20 mM ammonium bicarbonate in 20 min) to afford 0.050 g (15%) of the title compound as an off-white solid. LC-MS (ESI,
m/
z): 378 [M + H]
+.
1H NMR (400 MHz, CD
3OD) δ 9.46 (s, 2H), 9.33 (s, 1H), 8.16 (dd,
J = 1.5, 7.8 Hz, 1H), 7.64 (m, 1H), 7.10–7.24 (m, 2H), 5.11 (m, 1H), 3.16 (m, 1H), 2.84–3.04 (m, 2H), 2.69 (m, 2H), 2.39 (m, 1H), 2.13 (m, 1H), 1.89 (m, 2H), 1.71 (m, 2H), 1.39–1.64 (m, 4H) (
Figure S20).
13C NMR (150 MHz, DMSO-
d6) δ 175.78, 163.96, 160.47, 156.72, 155.31, 135.21, 131.64, 121.45, 120.93, 114.82, 112.49, 77.78, 66.17, 59.11, 51.69, 31.87, 31.58, 31.41, 23.71 (
Figure S21).
3.1.20. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-pyridazin-4-yl-1,2,4-oxadiazole (25)
A mixture of pyridazine-4-carbonitrile (1.0 g, 9.52 mmol, 1 eq), hydroxylamine hydrochloride (0.992 g, 14.3 mmol, 1.5 eq) and DIPEA (3.3 mL, 19.0 mmol, 2 eq) in EtOH (40 mL) was heated at 90 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc and water. The phases were separated. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of THF (0–70%) in hexane to afford 0.300 g (23%) of N-hydroxypyridazine-4-carboximidamide as an off-white solid. LC-MS (ESI, m/z): 139 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.45 (s, 1H), 9.25 (d, J = 6.1 Hz, 1H), 7.86 (m, 1H), 6.19 (br. s., 2H).
The title compound was prepared similarly to 24 using
N-hydroxypyridazine-4-carboximidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 378 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 9.81 (s, 1H), 9.53 (d,
J = 4.9 Hz, 1H), 8.27 (d,
J = 4.9 Hz, 1H), 8.14 (d,
J = 7.8 Hz, 1H), 7.69 (t,
J = 7.8 Hz, 1H), 7.26 (d,
J = 7.8 Hz, 1H), 7.19 (m, 1H), 5.08 (m, 1H), 3.02 (m, 1H), 2.70 (m, 2H), 2.41–2.62 (m, 2H), 2.33 (m, 1H), 1.90 (m, 1H), 1.73 (m, 2H), 1.61 (m, 2H), 1.32–1.55 (m, 4H) (
Figure S22).
13C NMR (150 MHz, DMSO-
d6) δ 176.17, 164.64, 156.77, 152.60, 148.23, 135.34, 131.67, 125.08, 123.92, 120.96, 114.83, 112.37, 77.81, 66.15, 59.10, 51.69, 31.87, 31.59, 31.41, 23.71 (
Figure S23).
3.1.21. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-phenyl-1,2,4-oxadiazole (26)
The title compound was prepared similarly to 23 using
N-hydroxybenzimidamide instead of
N-hydroxynicotinimidamide. LC-MS (ESI,
m/
z): 376 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.10 (m, 3H), 7.51–7.80 (m, 4H), 7.24 (d,
J = 8.8 Hz, 1H), 7.17 (t,
J = 7.6 Hz, 1H), 5.06 (m, 1H), 3.01 (m, 1H), 2.64–2.84 (m, 2H), 2.43–2.55 (m, 2H), 2.31 (m, 1H), 1.89 (m, 1H), 1.73 (m, 2H), 1.60 (m, 2H), 1.32–1.54 (m, 4H) (
Figure S24).
13C NMR (150 MHz, DMSO-
d6) δ 175.14, 167.59, 156.60, 134.78, 131.60, 131.50, 129.32, 127.12, 126.48, 120.86, 114.78, 113.03, 77.73, 66.14, 59.10, 51.67, 31.88, 31.57, 31.40, 23.70 (
Figure S25).
3.1.22. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(6-methoxy-3-pyridyl)-1,2,4-oxadiazole (27)
A mixture of 6-methoxynicotinonitrile (1.0 g, 7.45 mmol, 1 eq), hydroxylamine hydrochloride (0.777 g, 11.2 mmol, 1.5 eq) and K2CO3 (3.09 g, 22.4 mmol, 3 eq) in EtOH (50 mL) was heated at 80 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc and water. The phases were separated. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of THF (0–40%) in hexane to afford 0.200 g (16%) of N-hydroxy-6-methoxynicotinimidamide as an off-white solid. LC-MS (ESI, m/z): 168 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.43 (s, 1H), 7.93 (m, 1H), 6.83 (d, J = 8.4 Hz, 1H), 5.86 (br. s., 2H), 3.86 (s, 3H).
The title compound was prepared similarly to
24 using
N-hydroxy-6-methoxynicotinimidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 407 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.88 (d,
J = 2.0 Hz, 1H), 8.32 (dd,
J = 2.0, 8.5 Hz, 1H), 8.09 (d,
J = 7.5 Hz, 1H), 7.66 (t,
J = 7.8 Hz, 1H), 7.24 (d,
J = 7.8 Hz, 1H), 7.17 (t,
J = 7.5 Hz, 1H), 7.04 (d,
J = 8.5 Hz, 1H), 5.06 (m, 1H), 3.96 (s, 3H), 3.02 (m, 1H), 2.70 (m, 2H), 2.41–2.62 (m, 2H), 2.26–2.37 (m, 1H), 1.88 (m, 1H), 1.73 (m, 2H), 1.61 (m, 2H), 1.33–1.55 (m, 4H) (
Figure S26).
3.1.23. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-[5-(trifluoromethyl)-3-pyridyl]-1,2,4-oxadiazole (28)
A mixture of 5-(trifluoromethyl)nicotinonitrile (0.250 g, 1.45 mmol, 1 eq), hydroxylamine hydrochloride (0.151 g, 2.18 mmol, 1.5 eq) and DIPEA (0.600 mL, 3.6 mmol) in EtOH (5 mL) was heated at 90 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc and water. The phases were separated. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was triturated with EtOAc. The white solid was filtered and dried under high vacuum to afford 0.250 g (83%) of N-hydroxy-5-(trifluoromethyl)nicotinimidamide. LC-MS (ESI, m/z): 206 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.15 (s, 1H), 8.96 (s, 1H), 8.36 (s, 1H), 6.18 (br. s., 2H).
The title compound was prepared similarly to
24 using
N-hydroxy-5-(trifluoromethyl)nicotinimidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 445 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 9.53 (s, 1H), 9.25 (s, 1H), 8.70 (s, 1H), 8.15 (d,
J = 7.3 Hz, 1H), 7.68 (t,
J = 7.8 Hz, 1H), 7.26 (d,
J = 7.8 Hz, 1H), 7.19 (t,
J = 7.3 Hz, 1H), 5.08 (m, 1H), 3.03 (m, 1H), 2.65–2.77 (m, 2H), 2.41–2.62 (m, 2H), 2.33 (m, 1H), 1.89 (m, 1H), 1.73 (m, 2H), 1.60 (m, 2H), 1.31–1.55 (m, 4H) (
Figure S27).
13C NMR (150 MHz, DMSO-
d6) δ 175.76, 164.91, 156.73, 151.54, 148.85, 135.23, 131.75, 131.65, 123.17, 120.93, 114.83, 112.47, 77.79, 66.19, 59.14, 51.70, 31.89, 31.58, 31.41, 23.69 (
Figure S28).
3.1.24. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-[4-(trifluoromethyl)-3-pyridyl]-1,2,4-oxadiazole (29)
A mixture of 4-(trifluoromethyl)nicotinonitrile (0.250 g, 1.45 mmol, 1 eq), hydroxylamine hydrochloride (0.151 g, 2.18 mmol, 1.5 eq) and DIPEA (0.506 mL, 2.91 mmol, 2 eq) in EtOH (10 mL) was heated at 90 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc and water. The phases were separated. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of THF (0–30%) in hexane to afford 0.140 g (47%) of N-hydroxy-4-(trifluoromethyl)nicotinimidamide as an off-white solid. LC-MS (ESI, m/z): 206 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.85 (d, J = 4.9 Hz, 1H), 8.75 (s, 1H), 7.78 (d, J = 4.9 Hz, 1H), 6.04 (br. s., 2H).
The title compound was prepared similarly to
24 using
N-hydroxy-4-(trifluoromethyl)nicotinimidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 445 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 9.22 (s, 1H), 9.11 (d,
J = 4.5 Hz, 1H), 8.06 (m, 2H), 7.68 (t,
J = 7.3 Hz, 1H), 7.26 (d,
J = 8.3 Hz, 1H), 7.18 (t,
J = 7.3 Hz, 1H), 5.08 (m, 1H), 3.03 (m, 1H), 2.70 (m, 2H), 2.41–2.62 (m, 2H), 2.33 (m, 1H), 1.89 (m, 1H), 1.72 (m, 2H), 1.60 (m, 2H), 1.30–1.54 (m, 4H) (
Figure S29).
13C NMR (150 MHz, DMSO-
d6) δ 175.38, 164.73, 156.73, 153.66, 151.87, 135.23, 131.57, 125.08, 120.94, 120.73, 120.70, 119.84, 114.79, 112.34, 77.75, 66.12, 59.02, 51.65, 31.82, 31.52, 31.37, 23.67 (
Figure S30).
3.1.25. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(4-methyl-3-pyridyl)-1,2,4-oxadiazole (30)
A mixture of 4-methylnicotinonitrile (0.500 g, 4.23 mmol, 1 eq), hydroxylamine hydrochloride (0.441 g, 6.35 mmol, 1.5 eq) and NEt3 (1.2 mL, 8.47 mmol, 2 eq) in EtOH (10 mL) was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of THF (0–40%) in hexane to afford 0.375 g (58%) of N-hydroxy-4-methylnicotinimidamide as an off-white solid. LC-MS (ESI, m/z): 152 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.41 (m, 2H), 7.26 (d, J = 5.0 Hz, 1H), 5.89 (br. s., 2H), 2.36 (s, 3H).
The title compound was prepared similarly to
24 using
N-hydroxy-4-methylnicotinimidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 391 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 9.11 (s, 1H), 8.62 (d,
J = 5.4 Hz, 1H), 8.11 (d,
J = 7.8 Hz, 1H), 7.66 (t,
J = 8.3 Hz, 1H), 7.49 (d,
J = 5.4 Hz, 1H), 7.25 (d,
J = 8.3 Hz, 1H), 7.17 (t,
J = 7.8 Hz, 1H), 5.07 (m, 1H), 3.02 (m, 1H), 2.71 (m, 2H), 2.65 (s, 3H), 2.41–2.62 (m, 2H), 2.33 (m, 1H), 1.89 (m, 1H), 1.72 (m, 2H), 1.60 (m, 2H), 1.31–1.54 (m, 4H) (
Figure S31).
13C NMR (150 MHz, DMSO-
d6) δ 174.44, 166.42, 156.62, 151.34, 149.79, 146.79, 134.91, 131.59, 126.30, 122.42, 120.87, 120.70, 114.74, 112.73, 77.68, 66.16, 59.10, 51.70, 31.87, 31.56, 31.41, 23.69, 20.99 (
Figure S32).
3.1.26. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(3-methyl-4-pyridyl)-1,2,4-oxadiazole (31)
A mixture of 3-methylisonicotinonitrile (0.250 g, 2.11 mmol, 1 eq), hydroxylamine hydrochloride (0.294 g, 4.24 mmol, 2 eq) and NaHCO3 (0.712 g, 8.48 mmol, 4 eq) in MeOH (5 mL) was heated at 70 °C for 6 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (YMC-Actus C18 column, 250 × 20 mm, 5 µ; 5% MeOH in 20 mM ammonium bicarbonate) to afford 0.180 g (56%) of N-hydroxy-3-methylisonicotinimidamide as an off-white solid. LC-MS (ESI, m/z): 152 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.47 (s, 1H), 8.40 (d, J = 4.8 Hz, 1H), 7.29 (d, J = 4.8 Hz, 1H), 5.88 (br. s., 2H), 2.35 (s, 3H).
The title compound was prepared similarly to
24 using
N-hydroxy-3-methylisonicotinimidamide instead of
N-hydroxypyrimidine-5-carboximidamide. LC-MS (ESI,
m/
z): 391 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.70 (s, 1H), 8.64 (d,
J = 5.0 Hz, 1H), 8.11 (d,
J = 7.5 Hz, 1H), 7.96 (d,
J = 5.0 Hz, 1H), 7.67 (t,
J = 7. 8 Hz, 1H), 7.26 (d,
J = 7.5 Hz, 1H), 7.18 (t,
J = 7.5 Hz, 1H), 5.08 (m, 1H), 3.03 (m, 1H), 2.71 (m, 2H), 2.63 (s, 3H), 2.41–2.62 (m, 2H), 2.33 (m, 1H), 1.90 (m, 1H), 1.74 (m, 2H), 1.61 (m, 2H), 1.31–1.55 (m, 4H) (
Figure S33).
13C NMR (150 MHz, DMSO-
d6) δ 174.72, 166.87, 156.62, 152.39, 147.93, 135.03, 132.86, 131.85, 131.60, 122.75, 120.93, 114.78, 112.63, 77.66, 66.15, 59.04, 51.69, 31.83, 31.50, 31.34, 23.69, 18.49 (
Figure S34).
3.1.27. 2-((1-Cyclopentylpyrrolidin-3-yl)oxy)benzonitrile (33)
To a solution of 19 (1.6 g, 5.53 mmol, 1 eq) in dioxane (5 mL) was added 25% NH4OH (10 mL; 79.3 mmol, 14 eq). The reaction mixture was heated at 100 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between 10% LiOH and 10% MeOH in CH2Cl2. The phases were separated. The aqueous phase was extracted with 10% MeOH in CH2Cl2. The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford 0.900 g of 2-((1-cyclopentylpyrrolidin-3-yl)oxy)benzamide 32, which was used in the next step without further purification.
To a solution of 32 (0.900 g, 3.28 mmol) in EtOAc (20 mL) was added 50% T3P in EtOAc (4.8 mL, 16.4 mmol, 5 eq). The reaction mixture was heated at 60 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between water and 10% MeOH in CH2Cl2. The phases were separated. The aqueous phase was extracted twice with 10% MeOH in CH2Cl2. The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of MeOH (0–5%) in CH2Cl2 to afford 0.800 g (54% over two steps) of the title compound as a brown oil. LC-MS (ESI, m/z): 257 [M + H]+.
3.1.28. 3-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-5-(4-pyridyl)-1,2,4-oxadiazole (35)
A mixture of 2-((1-cyclopentylpyrrolidin-3-yl)oxy)benzonitrile (0.700 g, 2.73 mmol, 1 eq), hydroxylamine hydrochloride (0.569 g, 8.19 mmol, 3 eq) and DIPEA (1.9 mL, 10.9 mmol, 4 eq) in EtOH (10 mL) was heated at 70 °C overnight. After cooling to room temperature, the reaction mixture was diluted with water and extracted with 10% MeOH in CH2Cl2 (3×). The organic phases were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford 0.700 g (89%) of 2-((1-cyclopentylpyrrolidin-3-yl)oxy)-N-hydroxybenzimidamide 34 as an off-white solid, which was used in the next step without further purification. LC-MS (ESI, m/z): 290.
To a solution of
34 (0.700 g, 2.42 mmol, 1 eq) in DMF (10 mL) were added isonicotinic acid (0.596 g, 4.84 mmol, 2 eq), EDC.HCl (0.927 g, 4.84 mmol, 2 eq), HOBt (0.654 g, 4.84 mmol, 2 eq) and DIPEA (1.7 mL, 9.68 mmol, 4 eq). The reaction mixture was stirred at room temperature for 16 h and was heated at 80 °C for 16 h. After cooling to room temperature, the reaction mixture was partitioned between water and EtOAc. The phases were separated. The organic phase was washed with brine, dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Xbridge C18 column, 250 × 19 mm, 10µ; 30 to 75% ACN in 20 mM ammonium bicarbonate in 22 min) to afford 0.080 g (9%) of the title compound as a white solid. LC-MS (ESI,
m/
z): 377 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.91 (d,
J = 5.9 Hz, 2H), 8.10 (d,
J = 5.9 Hz, 2H), 7.98 (d,
J = 7.4 Hz, 1H), 7.57 (t,
J = 7.1 Hz, 1H), 7.15 (m, 2H), 5.01 (m, 1H), 3.02 (m, 1H), 2.65 (m, 2H), 2.42–2.55 (m, 2H), 2.27 (m, 1H), 1.86 (m, 1H), 1.70 (m, 2H), 1.59 (m, 2H), 1.29–1.53 (m, 4H) (
Figure S35).
13C NMR (150 MHz, DMSO-
d6) δ 172.53, 167.26, 156.23, 151.25, 132.99, 131.10, 130.54, 121.35, 120.62, 115.40, 114.33, 77.46, 66.15, 59.07, 51.65, 31.85, 31.55, 31.40, 23.69 (
Figure S36).
3.1.29. 2-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-5-(4-pyridyl)-1,3,4-oxadiazole (38)
To a solution of
36 (0.150 g, 0.545 mmol, 1 eq) in POCl
3 (5 mL) was added isonicotinohydrazide (0.075 g, 0.545 mmol, 1 eq). The reaction mixture was heated at 90 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was taken up with sat. NaHCO
3 and was extracted twice with 5% MeOH in CH
2Cl
2. The organic phases were combined, washed with brine, dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (YMC-Actus C18 column, 250 × 20 mm, 5µ; 30 to 75% ACN in 20 mM ammonium bicarbonate in 20 min) to afford 0.040 g (19%) of the title compound as yellow solid. LC-MS (ESI,
m/
z): 377 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.85 (m, 2H), 8.01 (m, 3H), 7.62 (t,
J = 7.3 Hz, 1H), 7.24 (d,
J = 8.3 Hz, 1H), 7.16 (t,
J = 7.3 Hz, 1H), 5.07 (m, 1H), 2.91 (m, 1H), 2.79 (m, 2H), 2.41–2.62 (m, 2H), 2.34 (m, 1H), 1.88 (m, 1H), 1.72 (m, 2H), 1.28–1.65 (m, 6H) (
Figure S37).
13C NMR (150 MHz, DMSO-
d6) δ 164.10, 162.43, 155.82, 151.02, 133.97, 130.70, 130.55, 120.87, 120.12, 114.65, 112.52, 77.58, 66.24, 59.31, 51.83, 31.91, 31.60, 31.40, 23.67 (
Figure S38).
3.1.30. 3-Bromo-5-(2-methoxyphenyl)-1,2,4-thiadiazole (41)
A mixture of 3-bromo-5-chloro-1,2,4-thiadiazole (1.0 g, 5.01 mmol, 1 eq), (2-methoxyphenyl)boronic acid (0.381 g, 2.51 mmol, 0.5 eq) and CsF (1.52 g, 10.0 mmol, 2 eq) in dioxane (20 mL) and water (5 mL) was degassed with Ar for 30 min. Pd(dppf)Cl2 (0.367 g, 0.501 mmol, 0.1 eq) was added and the reaction mixture was heated at 85 °C overnight. After cooling to room temperature, the reaction mixture was diluted with EtOAc and filtered. The phases of the filtrate were separated. The organic phase was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (0–20%) in hexane to afford 0.620 g (46%) of the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 6.6 Hz, 1H), 7.69 (t, J = 7.2 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 4.13 (s, 3H).
3.1.31. 5-(2-Methoxyphenyl)-3-(pyridin-4-yl)-1,2,4-thiadiazole (42)
A mixture of 41 (0.550 g, 2.03 mmol, 1 eq), pyridin-4-ylboronic acid (0.374 g, 3.04 mmol, 1.5 eq) and K2CO3 (0.841 g, 6.09 mmol, 3 eq) in dioxane (20 mL) and water (5 mL) was degassed with Ar for 30 min. Pd(dppf)Cl2 (0.149 g, 0.203 mmol, 0.1 eq) was added and the reaction mixture was heated at 85 °C for 16 h. After cooling to room temperature, the reaction mixture was diluted with EtOAc and filtered. The phases of the filtrate were separated. The organic phase was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (0–40%) in hexane to afford 0.450 g (83%) of the title compound as a white solid. LC-MS (ESI, m/z): 270 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 5.9 Hz, 2H), 8.47 (dd, J = 1.4, 7.8 Hz, 1H), 8.21 (d, J = 6.0 Hz, 2H), 7.68 (m, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 4.16 (s, 3H).
3.1.32. (3-(Pyridin-4-yl)-1,2,4-thiadiazol-5-yl)phenol (43)
To a solution of 42 (0.350 g, 1.30 mmol, 1 eq) in NMP (8 mL) were added LiBr (1.13 g, 13.0 mmol, 10 eq) and pTsOH (1.12 g, 6.50 mmol, 5 eq). The reaction mixture was heated at 150 °C for 1.5 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with EtOAc (3×). The organic phases were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (0–40%) in hexane to afford 0.180 g (54%) of the title compound as a white solid. LC-MS (ESI, m/z): 256 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.99 (s, 1H), 8.79 (d, J = 4.6 Hz, 2H), 8.37 (d, J = 7.6 Hz, 1H), 8.22 (d, J = 4.4 Hz, 2H), 7.50 (t, J = 7.9 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.10 (m, J = 7.6 Hz, 1H).
3.1.33. tert-Butyl 3-(2-(3-(pyridin-4-yl)-1,2,4-thiadiazol-5-yl)phenoxy)pyrrolidine-1-carboxylate (44)
To a solution of 43 (0.180 g, 0.705 mmol, 1 eq) in THF (5 mL) were added PPh3 (0.277 g, 1.06 mmol, 1.5 eq) and 40% DIAD in toluene (0.520 mL, 1.058 mmol, 1.5 eq). The reaction mixture was stirred at room temperature for 10 min. tert-Butyl 3-hydroxypyrrolidine-1-carboxylate (0.158 g, 0.846 mmol, 1.2 eq) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc (0–50%) in hexane to afford 0.105 g (35%) of the title compound as a white solid. LC-MS (ESI, m/z): 425 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 5.7 Hz, 2H), 8.49 (d, J = 6.9 Hz, 1H), 8.20 (d, J = 5.9 Hz, 2H), 7.67 (t, J = 7.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 5.49 (m, 1H), 3.76 (m, 1H), 3.66 (m, 1H), 3.48 (m, 2H), 2.26 (m, 2H), 1.41 (s, 9H).
3.1.34. 3-(Pyridin-4-yl)-5-(2-(pyrrolidin-3-yloxy)phenyl)-1,2,4-thiadiazole dihydrochloride (45)
To a solution of 44 (0.105 g, 0.247 mmol, 1 eq) in dioxane (0.5 mL) cooled at 0 °C was added 4N HCl in dioxane (2.0 mL, 8.00 mmol, 32 eq). The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was triturated with Et2O. The white solid was filtered and dried under high vacuum to afford quantitatively the title compound. LC-MS (ESI, m/z): 325 [M + H]+.
3.1.35. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(4-pyridyl)-1,2,4-thiadiazole (46)
To a solution of
45 (0.080 g, 0.223 mmol, 1 eq) in CH
2Cl
2 (1 mL) and MeOH (1 mL) was added cyclopentanone (0.049 mL, 0.557 mmol, 2.5 eq) and the reaction mixture was stirred at room temperature for 2 h. NaBH(OAc)
3 (0.094 g, 0.446 mmol, 2 eq) was added portionwise and the reaction mixture was stirred at room temperature overnight. The reaction mixture was washed with water and the phases were separated. The aqueous phase was extracted with CH
2Cl
2 (3×). The organic phases were combined, dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Xterra C18 column,250 × 19 mm, 10µ; 20 to 60% ACN in 20 mM ammonium bicarbonate in 20 min) to afford 0.008 g (9%) of the title compound as a white solid. LC-MS (ESI,
m/
z): 393 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.80 (d,
J = 5.9 Hz, 2H), 8.49 (d,
J = 6.9 Hz, 1H), 8.22 (d,
J = 5.9 Hz, 2H), 7.65 (t,
J = 7.6 Hz, 1H), 7.33 (d,
J = 8.3 Hz, 1H), 7.25 (t,
J = 7.1 Hz, 1H), 5.33 (m, 1H), 2.76–3.12 (m, 4H), 2.30–2.61 (m, 2H), 2.01 (m., 1H), 1.78 (m, 2H), 1.65 (m, 2H), 1.37–1.58 (m, 4H) (
Figure S39).
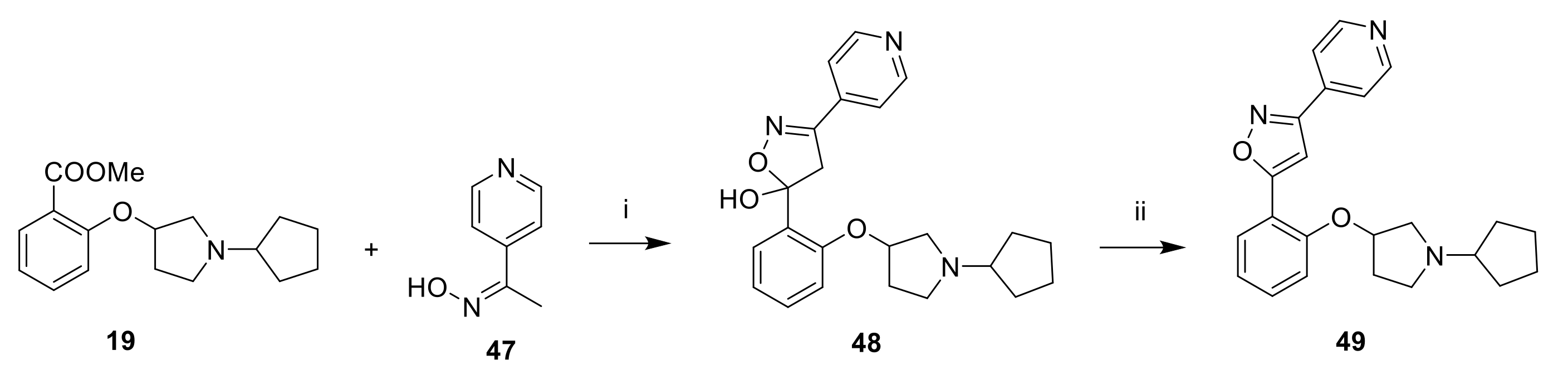
3.1.36. 5-[2-(1-Cyclopentylpyrrolidin-3-yl)oxyphenyl]-3-(4-pyridyl)-1,2,4-thiadiazole (49)
To a solution 4-acetylpyridine oxime (0.200 g, 1.47 mmol, 1 eq) in THF (3 mL) cooled at −78 °C was added dropwise 2M LDA in hexane (2.2 mL, 4.40 mmol, 3 eq) over 30 min. A solution of
19 (0.458 g, 1.62 mmol, 1.1 eq) in THF (1 mL) was added dropwise and the reaction mixture was stirred at −78 °C for 2 h. The reaction was quenched by the addition of sat. NH
4Cl. After warming to room temperature, the reaction mixture was extracted twice with EtOAc. The organic phases were combined, dried over Na
2SO
4, filtered and concentrated under reduced pressure to afford 5-(2-((1-cyclopentylpyrrolidin-3-yl)oxy)phenyl)-3-(pyridin-4-yl)-4,5-dihydroisoxazol-5-ol (48) which was dissolved in MeOH (3 mL). 12N HCl (3.0 mL, 36.0 mmol) was added and the reaction mixture was heated at 70 °C for 3 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Xterra RP18 column,250 × 19 mm, 10µ; 30 to 90% MeOH in 20 mM ammonium bicarbonate in 24 min) to afford 0.035 g (12%) of the title compound as a beige solid. LC-MS (ESI,
m/
z): 393 [M + H]
+.
1H NMR (400 MHz, DMSO-
d6) δ 8.77 (d,
J = 5.4 Hz, 2H), 7.93 (d,
J = 7.8 Hz, 1H), 7.85 (d,
J = 5.4 Hz, 2H), 7.47–7.57 (m, 2H), 7.22 (d,
J = 8.3 Hz, 1H), 7.14 (t,
J = 7.6 Hz, 1H), 5.13 (m, 1H), 2.75–2.99 (m, 3H), 2.47–2.57 (m, 1H), 2.41 (m, 1H), 2.31 (m, 1H), 1.91 (m, 1H), 1.77 (m, 2H), 1.61 (m, 2H), 1.34–1.55 (m, 4H) (
Figure S40).
13C NMR (150 MHz, DMSO-
d6) δ 167.09, 160.92, 154.25, 150.73, 136.04, 132.16, 127.33, 121.04, 120.92, 115.96, 114.42, 101.68, 77.59, 66.08, 59.28, 51.71, 31.80, 31.60, 31.51, 23.73 (
Figure S41).


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























