
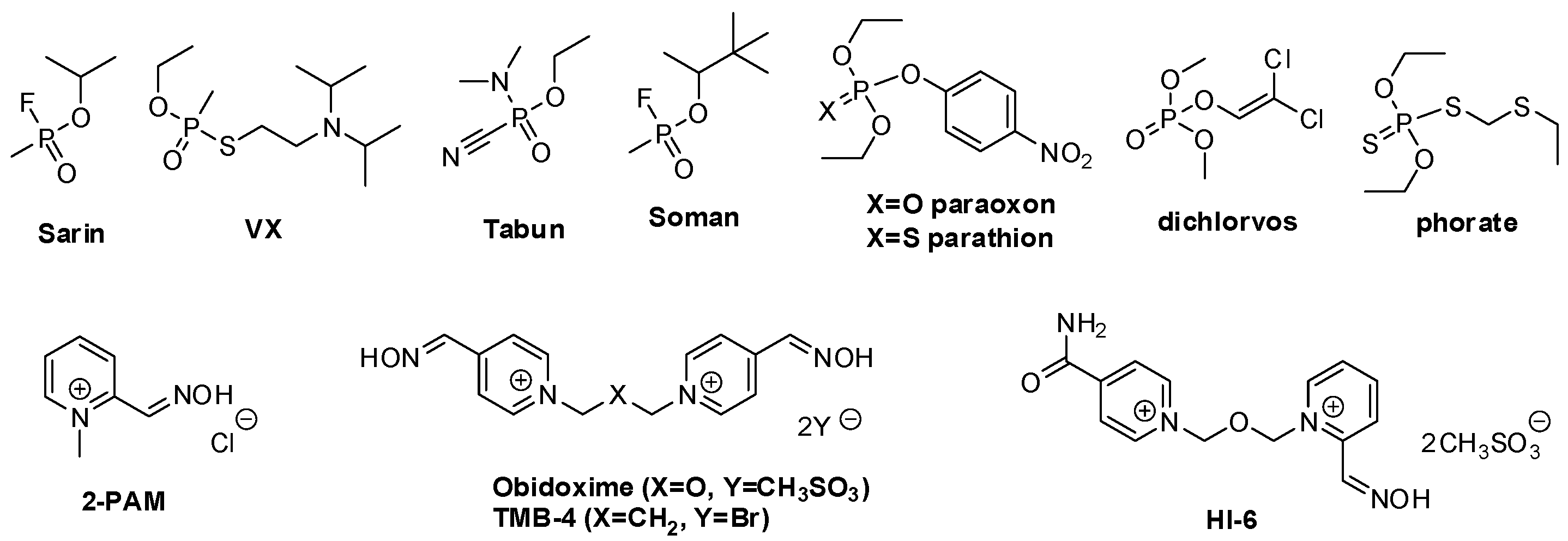
Discovery of Novel Non-Oxime Reactivators Showing In Vivo Antidotal Efficiency for Sarin Poisoned Mice

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
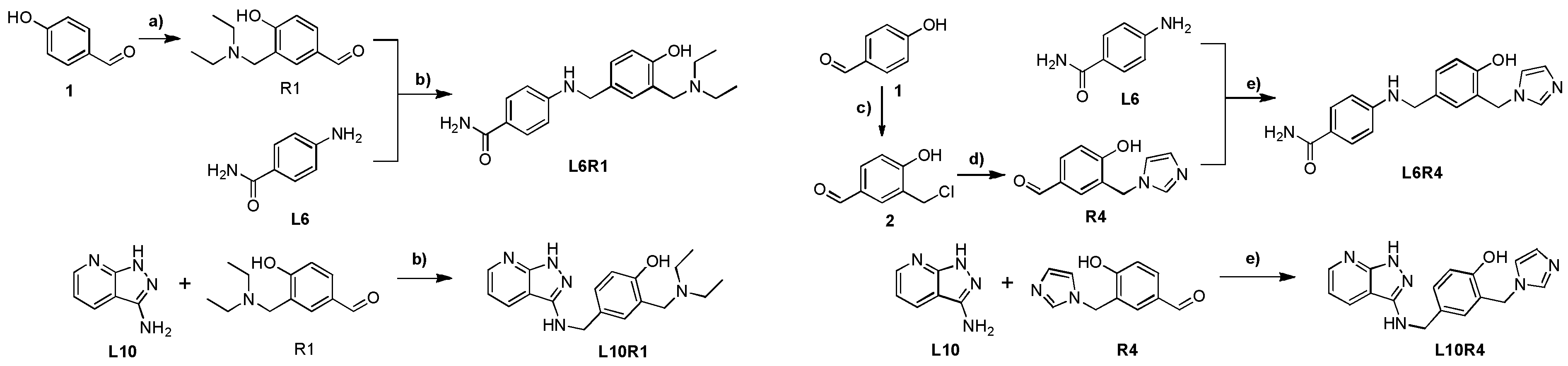
2.1. Synthesis
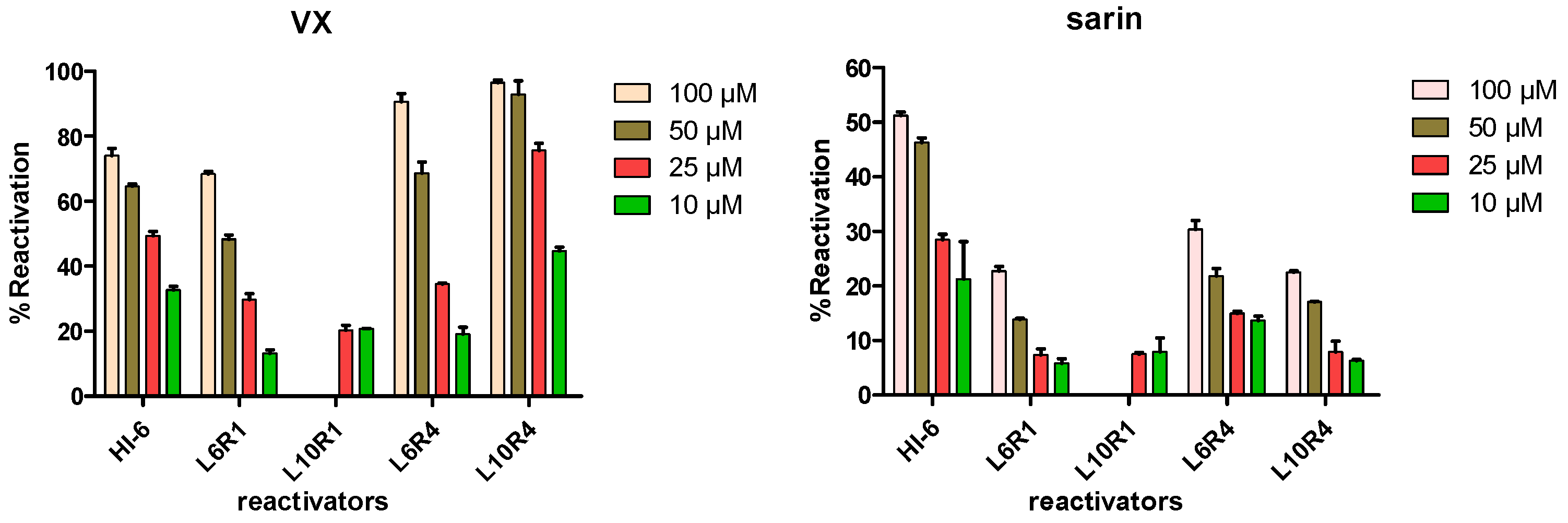
2.2. In Vitro Inhibition and Reactivation Experiments
2.3. Determination of Reactivation Kinetics
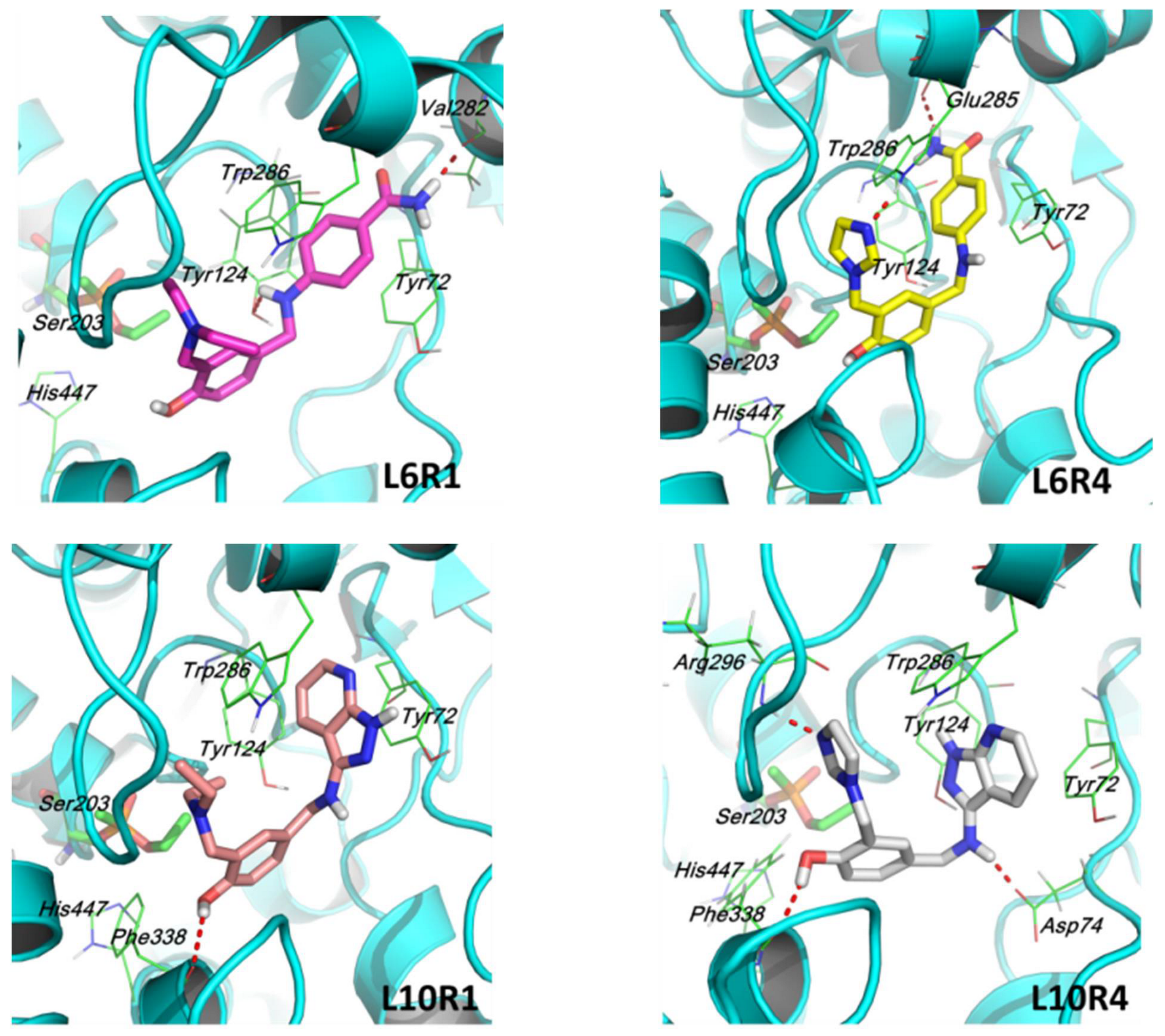
2.4. Molecular Docking Simulation
2.5. In Vivo Biological Experiments
3. Experimental Section
3.1. Chemicals
3.2. Synthesis Procedures for the Preparation of L6R1, L10R1, L6R4, and L10R4
3.3. Computational Methods
3.4. General In Vitro AChE Screening Information
3.5. Procedures of hAChE Inhibition Experiments
- (1)
- A stock solution of hAChE was diluted 2000-fold with PBS (0.1 M, pH = 7.4, 0.1% BSA);
- (2)
- To 20 μL of the diluted enzyme, 10 μL reactivator solutions (reactivator final concentrations: 10, 50, 200, 500, and 1000 μM, and each sample was measured in duplicate in parallel in a 96-well plate) were added, and the mixture was incubated for 15 min at 25 °C. A positive control was run in parallel by adding 10 μL of PBS instead of reactivator solution to the enzyme.
- (3)
- For each sample in 96-well plate, 30 μL of ATCh (3.0 mM, pH = 7.4 PBS), and 150 μL of DTNB (0.75 mM, pH = 7.0 PBS) were added. Then, the resulting mixture was centrifuged at 4 °C for 1 min to remove bubbles, and the reaction product was monitored immediately by testing the absorption value at 412 nm (0 < abs < 3).
3.6. Procedures of Reactivation Experiments
- (1)
- A stock solution of hAChE was diluted 2000-fold with PBS (0.1 M, pH = 7.4, 0.1% BSA); the concentrations of different nerve agents were determined by a pre-experiment similar to the inhibition experiment to attain an inhibition plateau from 90% to 95%. We tried carefully to control the dosage of OP used to avoid 100% inhibition of hAChE, which meant that all OP used had bound to the enzyme, and there was no OP presented in the reaction mixture. The final concentrations of OPs were as follows: VX, 3*107 fold diluted; sarin, 1.6*106 fold diluted.
- (2)
- The diluted hAChE (20 μL) was incubated with different nerve agents (10 μL) at 25 °C for 15 min. Then, the inhibited enzyme was incubated with reactivators (15 μL, 300/150/75/30 μM) at 25 °C for 30 min (final concentrations of reactivators were 100/50/25/10 μM).
- (3)
- For each sample in a 96-well plate, 30 μL of ATCh (3.0 mM, pH = 7.4 PBS) and 150 μL of DTNB (0.75 mM, pH = 7.0 PBS) were added. Then, the resulting mixture was centrifuged at 4 °C for 1 min to remove bubbles, and the reaction product was monitored immediately by testing the absorption value at 412 nm (0 < abs < 2). Blank samples were run in parallel and consisted of: (a) a positive control (P): an uninhibited enzyme (20 μL) was used instead of the inhibited enzyme; (b) a negative control (N): PBS (25 μL, 0.1 M, pH 7.4) was used instead of reactivators. %Reactivation was calculated using the formula: %Reactivation = 100*(S-N)/(P-N).
3.7. Determination of Reactivation Kinetics

3.8. Details of the In Vivo Reactivation Experiments
- Animals were pretreated with L10R1, L6R4, and L10R4 at a dose of 60 mg/kg (ip); in parallel, two sets of mice pretreated with isotonic saline alone were challenged with 2*LD50 dose of sarin (85 μg/Kg), and one set of the mice was treated 1 min later with atropine sulfate (0.5 mg/Kg, control 2 in Table 2).
- Mice were observed for neurological toxicity symptoms such as muscles twitching, seizures, and convulsions after sarin or antidote administration, and the 48 h survival was finally recorded.
- Mice were pretreated with different antidotes (including 2-PAM, HI-6, L10R1, L6R4, and L10R4);
- 15 min later, a 2*LD50 dose of sarin (85 μg/Kg) was administrated (ip), and atropine sulfate (0.5 mg/Kg) was administrated 1 min later.
- Mice were observed for neurological toxicity symptoms such as muscles twitching, seizures, and convulsions after sarin or antidote administration, and the 48 h survival was finally recorded.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeyaratnam, J. Acute Pesticide Poisoning: A Major Global Health Problem. World Health Stat. Q. 1990, 43, 139–144. [Google Scholar] [PubMed]
- Marrs, T.C. Organophosphate Poisoning. Pharmacol. Ther. 1993, 58, 51. [Google Scholar] [CrossRef]
- Tu, A.T. Toxicological and Chemical Aspects of Sarin Terrorism in Japan in 1994 and 1995. Toxin Rev. 2007, 26, 231. [Google Scholar] [CrossRef]
- Eddleston, M.; Buckley, N.A.; Eyer, P.; Dawson, A.H. Management of Acute Organophosphorus Pesticide Poisoning. Lancet 2008, 371, 2170–2171. [Google Scholar] [CrossRef]
- Jokanović, M.; Stojiljković, M.P. Current Understanding of the Application of Pyridinium Oximes as Cholinesterase Reactivators in Treatment of Organophosphate Poisoning. Eur. J. Pharmacol. 2006, 553, 10–17. [Google Scholar] [CrossRef]
- Jokanovic, M. Pyridinium Oximes as Cholinesterase Reactivators. Structure-Activity Relationship and Efficacy in the Treatment of Poisoning with Organophosphorus Compounds. Curr. Med. Chem. 2009, 16, 2177–2188. [Google Scholar] [CrossRef]
- Jokanović, M. Medical Treatment of Acute Poisoning with Organophosphorus and Carbamate Pesticides. Toxicol. Lett. 2009, 190, 107–115. [Google Scholar] [CrossRef]
- Bajgar, J. Organophosphates/Nerve Agent Poisoning: Mechanism of Action, Diagnosis, Prophylaxis, and Treatment. Adv. Clin. Chem. 2004, 38, 151–216. [Google Scholar] [CrossRef]
- Kassa, J. Review of Oximes in the Antidotal Treatment of Poisoning by Organophosphorus Nerve Agents. J. Toxicol. Clin. Toxicol. 2002, 40, 803–816. [Google Scholar] [CrossRef]
- Shih, T.-M.; Skovira, J.W.; O’Donnell, J.; McDonough, J.H. In Vivo Reactivation by Oximes of Inhibited Blood, Brain and Peripheral Tissue Cholinesterase Activity Following Exposure to Nerve Agents in Guinea Pigs. Chem. Interact. 2010, 187, 207–214. [Google Scholar] [CrossRef]
- Little, P.J.; Scimeca, J.A.; Martin, B.R. Distribution of [3H]Diisopropyl-Fluorophosphate, [3H]Soman, [3H]Sarin, and their Metabolites in Mouse Brain. Drug Metab. Dispos. 1988, 16, 515–520. [Google Scholar] [PubMed]
- Rutland, J.P. The Effect of Some Oximes in Sarin Poisoning. Br. J. Pharmacol. Chemother. 1958, 13, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, T.-M.; Skovira, J.W.; O’Donnell, J.C.; McDonough, J.H. Treatment with Tertiary Oximes Prevents Seizures and Improves Survival Following Sarin Intoxication. J. Mol. Neurosci. 2009, 40, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kalisiak, J.; Ralph, E.C.; Zhang, J.; Cashman, J.R. Amidine−Oximes: Reactivators for Organophosphate Exposure. J. Med. Chem. 2011, 54, 3319–3330. [Google Scholar] [CrossRef]
- Kalisiak, J.; Ralph, E.C.; Cashman, J.R. Nonquaternary Reactivators for Organophosphate-Inhibited Cholinesterases. J. Med. Chem. 2011, 55, 465–474. [Google Scholar] [CrossRef] [PubMed]
- de Koning, M.; Joosen, M.; Noort, D.; van Zuylen, A.; Tromp, M. Peripheral Site Ligand–Oxime Conjugates: A Novel Concept towards Reactivation of Nerve Agent-Inhibited Human Acetylcholinesterase. Bioorganic Med. Chem. 2011, 19, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Mercey, G.; Verdelet, T.; Saint-André, G.; Gillon, E.; Wagner, A.; Baati, R.; Jean, L.; Nachon, F.; Renard, P.-Y. First Efficient Uncharged Reactivators for the Dephosphylation of Poisoned Human Acetylcholinesterase. Chem. Commun. 2011, 47, 5295–5297. [Google Scholar] [CrossRef] [PubMed]
- Mercey, G.; Renou, J.; Verdelet, T.; Kliachyna, M.; Baati, R.; Gillon, E.; Arboléas, M.; Loiodice, M.; Nachon, F.; Jean, L.; et al. Phenyltetrahydroisoquinoline–Pyridinaldoxime Conjugates as Efficient Uncharged Reactivators for the Dephosphylation of Inhibited Human Acetylcholinesterase. J. Med. Chem. 2012, 55, 10791–10795. [Google Scholar] [CrossRef]
- Renou, J.; Loiodice, M.; Arboléas, M.; Baati, R.; Jean, L.; Nachon, F.; Renard, P.-Y. Tryptoline-3-Hydroxypyridinaldoxime Conjugates as Efficient Reactivators of Phosphylated Human Acetyl and Butyrylcholinesterases. Chem. Commun. 2014, 50, 3947–3950. [Google Scholar] [CrossRef]
- Kliachyna, M.; Santoni, G.; Nussbaum, V.; Renou, J.; Sanson, B.; Colletier, J.-P.; Arboléas, M.; Loiodice, M.; Weik, M.; Jean, L.; et al. Design, Synthesis and Biological Evaluation of Novel Tetrahydroacridine Pyridine-Aldoxime and-Amidoxime Hybrids as Efficient Uncharged Reactivators of Nerve Agent-Inhibited Human Acetylcholinesterase. Eur. J. Med. Chem. 2014, 78, 455–467. [Google Scholar] [CrossRef]
- McHardy, S.F.; Bohmann, J.A.; Corbett, M.R.; Campos, B.; Tidwell, M.W.; Thompson, P.M.; Bemben, C.J.; Menchaca, T.A.; Reeves, T.E.; Cantrell, W.R.; et al. Design, Synthesis, and Characterization of Novel, Nonquaternary Reactivators of GF-Inhibited Human Acetylcholinesterase. Bioorganic Med. Chem. Lett. 2014, 24, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, Y.-Q.; Zhou, X.-B.; Luo, Y.; Huang, C.-Q.; Wang, Y.-A.; Zheng, Z.-B.; Li, S. New Efficient Imidazolium Aldoxime Reactivators for Nerve Agent-Inhibited Acetylcholinesterase. Bioorganic Med. Chem. Lett. 2014, 24, 5743–5748. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, Y.-Q.; Wang, S.-Z.; Yao, L.; Nie, H.-F.; Wang, Y.-A.; Liu, X.-Y.; Zheng, Z.-B.; Li, S. Conjugates of Salicylaldoximes and Peripheral Site Ligands: Novel Efficient Nonquaternary Reactivators for Nerve Agent-Inhibited Acetylcholinesterase. Bioorganic Med. Chem. 2017, 25, 4497–4505. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Bi, H.; Liu, Y.-Q.; Nie, H.-F.; Yao, L.; Wang, S.-Z.; Yang, J.; Wang, Y.-A.; Liu, X.; Zheng, Z.-B. Design, Synthesis and Evaluation of New Classes of Nonquaternary Reactivators for Acetylcholinesterase Inhibited by Organophosphates. Bioorganic Chem. 2018, 81, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Katz, F.S.; Pecic, S.; Tran, T.H.; Trakht, I.; Schneider, L.; Zhu, Z.; Ton-That, L.; Luzac, M.; Zlatanic, V.; Damera, S.; et al. Discovery of New Classes of Compounds that Reactivate Acetylcholinesterase Inhibited by Organophosphates. ChemBioChem 2015, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Cadieux, C.L.; Wang, H.; Zhang, Y.; Koenig, J.; Shih, T.-M.; McDonough, J.; Koh, J.; Cerasoli, D. Probing the Activity of a Non-Oxime Reactivator for Acetylcholinesterase Inhibited by Organophosphorus Nerve Agents. Chem. Interact. 2016, 259, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Katz, F.S.; Pecic, S.; Schneider, L.; Zhu, Z.; Hastings-Robinson, A.; Luzac, M.; Macdonald, J.; Landry, D.W.; Stojanovic, M.N. New Therapeutic Approaches and Novel Alternatives for Organophosphate Toxicity. Toxicol. Lett. 2018, 291, 1–10. [Google Scholar] [CrossRef]
- de Koning, M.C.; Horn, G.; Worek, F.; van Grol, M. Discovery of a Potent Non-Oxime Reactivator of Nerve Agent Inhibited Human Acetylcholinesterase. Eur. J. Med. Chem. 2018, 157, 151–160. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A New and Rapid Colorimetric Determination of AcetylchoLinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Bester, S.M.; Guelta, M.A.; Cheung, J.; Winemiller, M.D.; Bae, S.Y.; Myslinski, J.; Pegan, S.D.; Height, J.J. Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI-6. Chem. Res. Toxicol. 2018, 31, 1405–1417. [Google Scholar] [CrossRef]
- Dirnhuber, P.; French, M.C.; Green, D.M.; Leadbeater, L.; Stratton, J.A. The Protection of Primates against Soman Poisoning by Pretreatment with Pyridostigmine. J. Pharm. Pharmacol. 1979, 31, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, D.M.; Brecht, K.M.; Doctor, B.P.; Wolfe, A.D. Comparison of Antidote Protection against Soman by Pyridostigmine, HI-6 and Acetylcholinesterase. J. Pharmacol. Exp. Ther. 1993, 264, 1085–1089. [Google Scholar] [PubMed]
- Hsiao, L.Y.; Getzville, N.Y.; Hsiao, L.Y.Y.; Musallam, H.A. Damascus, Bis-Methylene Ether Pyridinium Compound Preparation. U.S. Patent 5130438, 14 July 1992. [Google Scholar]
- Eddolls, J.; Mccormack, P.; Hodgson, A. Process for the Manufacture of HI-6 Dimethanesulfonate. H.K. Patent 1137422, 34 March 2013. [Google Scholar]
- Worek, F.; Thiermann, H.; Szinicz, L.; Eyer, P. Kinetic Analysis of Interactions between Human Acetylcholinesterase, Structurally Different Organophosphorus Compounds and Oximes. Biochem. Pharmacol. 2004, 68, 2237–2248. [Google Scholar] [CrossRef]
- Kovarik, Z.; Radić, Z.; Berman, H.A.; Simeon-Rudolf, V.; Reiner, E.; Taylor, P. Mutant Cholinesterases Possessing Enhanced Capacity for Reactivation of Their Phosphonylated Conjugates. Biochemistry 2004, 43, 3222–3229. [Google Scholar] [CrossRef]
- Sit, R.K.; Fokin, V.V.; Amitai, G.; Sharpless, K.B.; Taylor, P.; Radić, Z. Imidazole Aldoximes Effective in Assisting Butyrylcholinesterase Catalysis of Organophosphate Detoxification. J. Med. Chem. 2014, 57, 1378–1389. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kr/10−3min−1 | KD/μM | kr2/mM−1min−1 | IC50 (μM) | pKa | ||||
|---|---|---|---|---|---|---|---|---|
| VX | sarin | VX | sarin | VX | sarin | |||
| HI-6 | 39.7 ± 4.6 | 14.4 ± 1.4 | 45.8 ± 14.8 | 16.2 ± 8.6 | 0.866 | 0.889 | 668 ± 61 | 7.0 ± 0.5 |
| L6R1 | 70.8 ± 17.4 | 10.8 ± 0.6 | 216 ± 86.2 | 163 ± 20.1 | 0.328 | 0.066 | 439 ± 18 | 11.9 ± 0.4 |
| L10R1 | 4.9 ± 0.6 | 1.62 ± 0.11 | 3.7 ± 1.4 | 5.31 ± 1.13 | 1.32 | 0.304 | 30.9 ± 0.4 | 11.6 ± 0.4 |
| L6R4 | 369 ± 154 | 13.6 ± 1.2 | 58.8 ± 61.4 | 94.8 ± 21.9 | 6.27 | 0.143 | 402 ± 19 | 9.9 ± 0.4 |
| L10R4 | - | 8.40 ± 0.80 | - | 73.8 ± 20.5 | 1.57 ± 0.07 | 0.113 | 483 ± 33 | 10.2 ± 0.4 |
| Experiment | Antidote (mg/Kg) a | μM/Kg | Atropine (mg/Kg) | Sarin (μg/Kg) | Survival (48 h) | LogBB | LogP |
|---|---|---|---|---|---|---|---|
| 1 | L10R1 (60 b) | 184 | - | - | 6/6 | - | - |
| L6R4 (60 b) | 186 | - | - | 6/6 | - | - | |
| L10R4 (60 b) | 188 | - | - | 6/6 | - | - | |
| Control 1 | - | - | 85 | 0/8 | - | - | |
| Control 2 | - | 0.5 | 85 | 1/8 | - | - | |
| 2 | 2-PAM (30) HI-6 (45) | 174 94 | 0.5 0.5 | 85 85 | 2/10 10/10 | −0.01 −2.0 | −3.04 −6.68 |
| L10R1 (30 b) | 92 | 0.5 | 85 | 10/10 | −0.06 | 2.51 | |
| L6R4 (30 b) | 93 | 0.5 | 85 | 3/10 | −0.60 | 1.32 | |
| L10R4 (30 b) | 94 | 0.5 | 85 | 2/10 | −0.78 | 1.25 |
| Reactivators | Sarin-hAChE | VX-hAChE |
|---|---|---|
| HI-6 | 20-50-100-200-400 | 10-25-50-100-200 |
| L6R1 | 10-20-50-100-200-400 | 5-10-25-50-100-200 |
| L6R4 | 10-20-50-100-200-400 | 5-10-25-50-100-200 |
| L10R1 | 1-2-5-10-20-50 | 0.5-1-2.5-5-10-25 |
| L10R4 | 10-20-50-100-200-400 | 5-10-25-50-100-200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Z.; Zhang, X.; Nie, H.; Yao, L.; Liu, Y.; Zheng, Z.; Ouyang, Q. Discovery of Novel Non-Oxime Reactivators Showing In Vivo Antidotal Efficiency for Sarin Poisoned Mice. Molecules 2022, 27, 1096. https://doi.org/10.3390/molecules27031096
Wei Z, Zhang X, Nie H, Yao L, Liu Y, Zheng Z, Ouyang Q. Discovery of Novel Non-Oxime Reactivators Showing In Vivo Antidotal Efficiency for Sarin Poisoned Mice. Molecules. 2022; 27(3):1096. https://doi.org/10.3390/molecules27031096
Chicago/Turabian StyleWei, Zhao, Xinlei Zhang, Huifang Nie, Lin Yao, Yanqin Liu, Zhibing Zheng, and Qin Ouyang. 2022. "Discovery of Novel Non-Oxime Reactivators Showing In Vivo Antidotal Efficiency for Sarin Poisoned Mice" Molecules 27, no. 3: 1096. https://doi.org/10.3390/molecules27031096
APA StyleWei, Z., Zhang, X., Nie, H., Yao, L., Liu, Y., Zheng, Z., & Ouyang, Q. (2022). Discovery of Novel Non-Oxime Reactivators Showing In Vivo Antidotal Efficiency for Sarin Poisoned Mice. Molecules, 27(3), 1096. https://doi.org/10.3390/molecules27031096