
π–π Stacking Interaction of Metal Phenoxyl Radical Complexes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
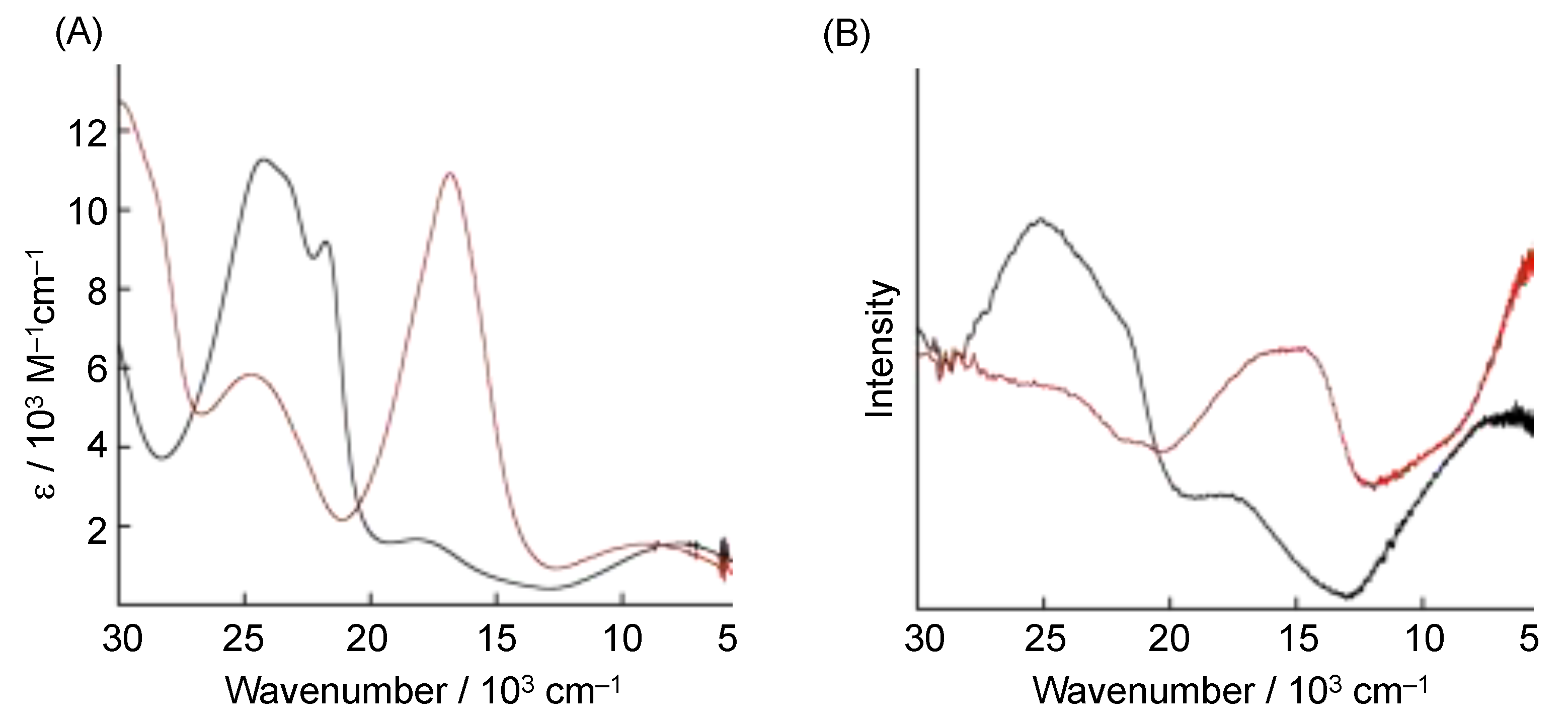{kind=link}
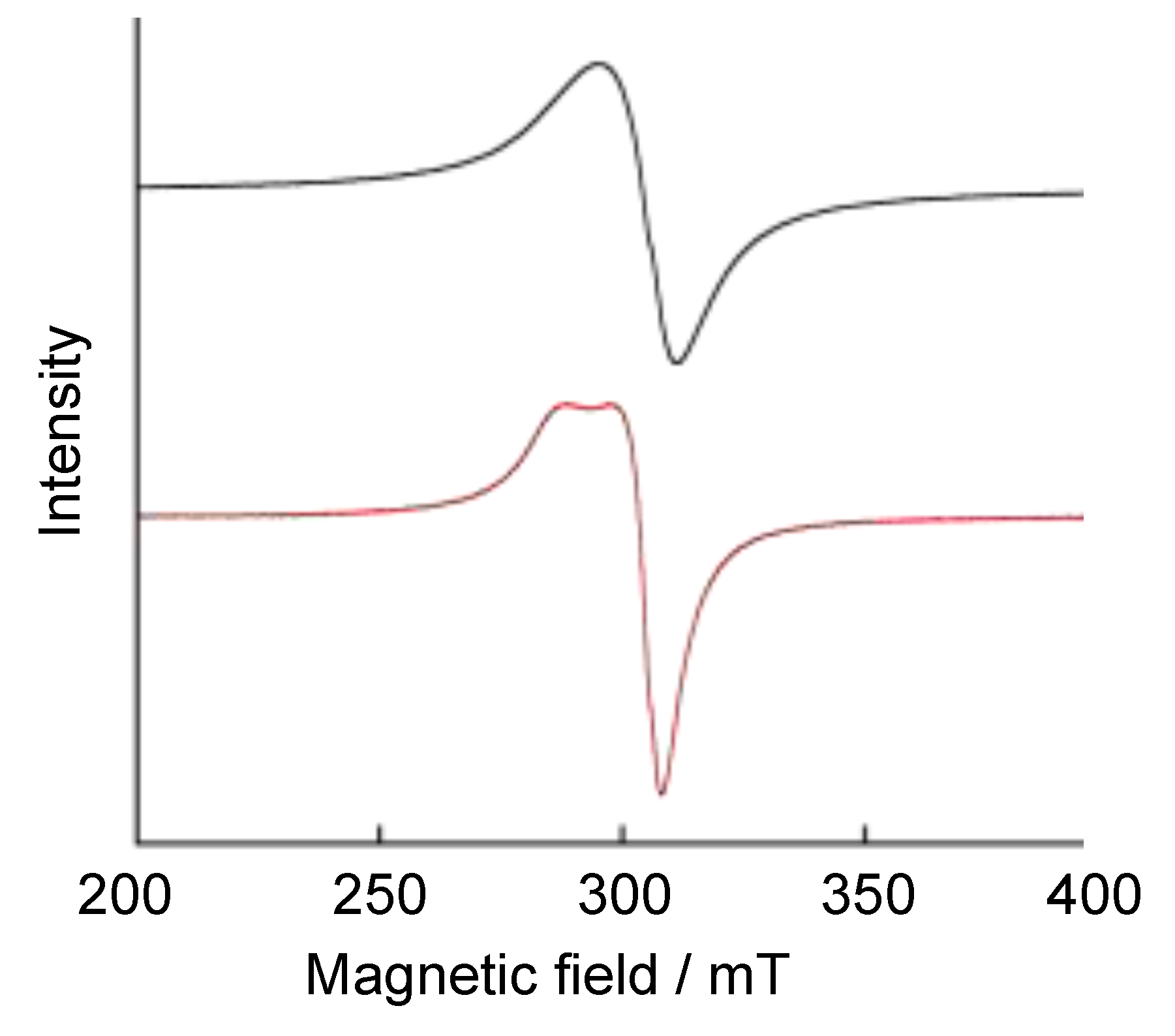{kind=link}
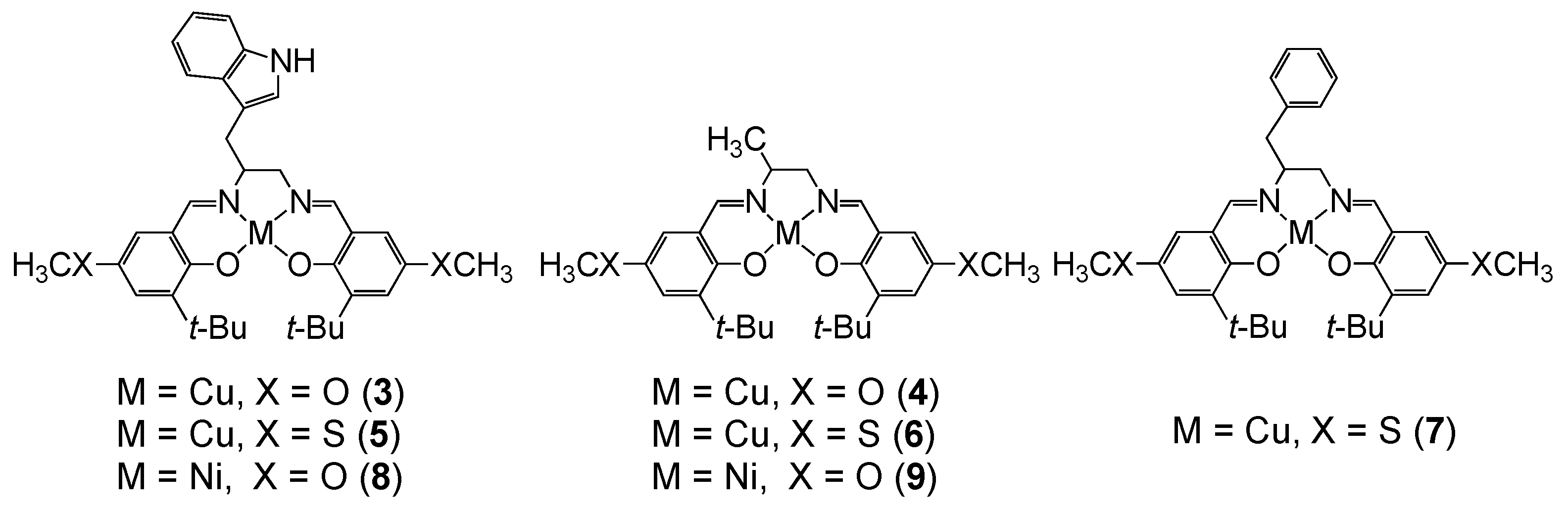{kind=link}
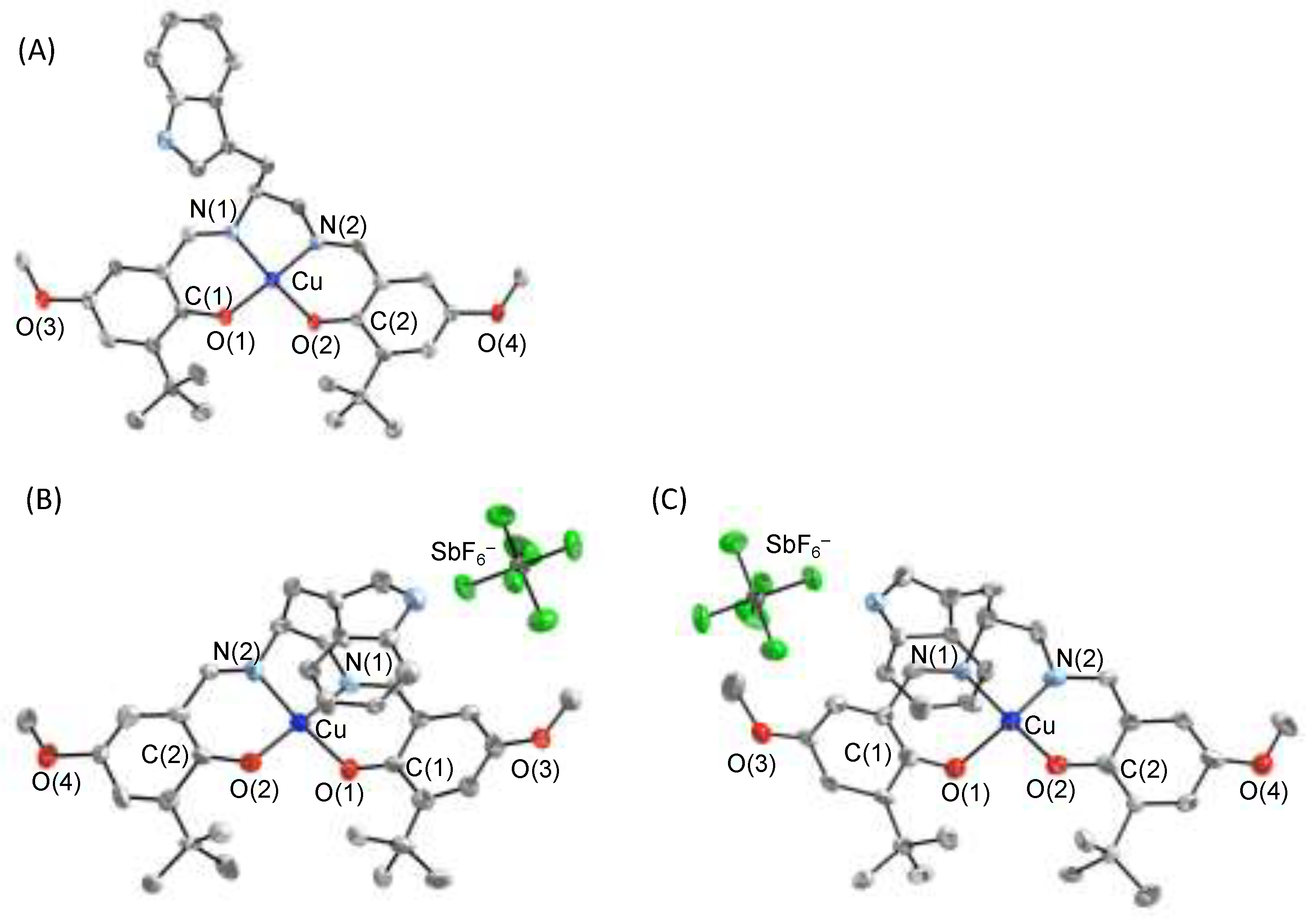{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
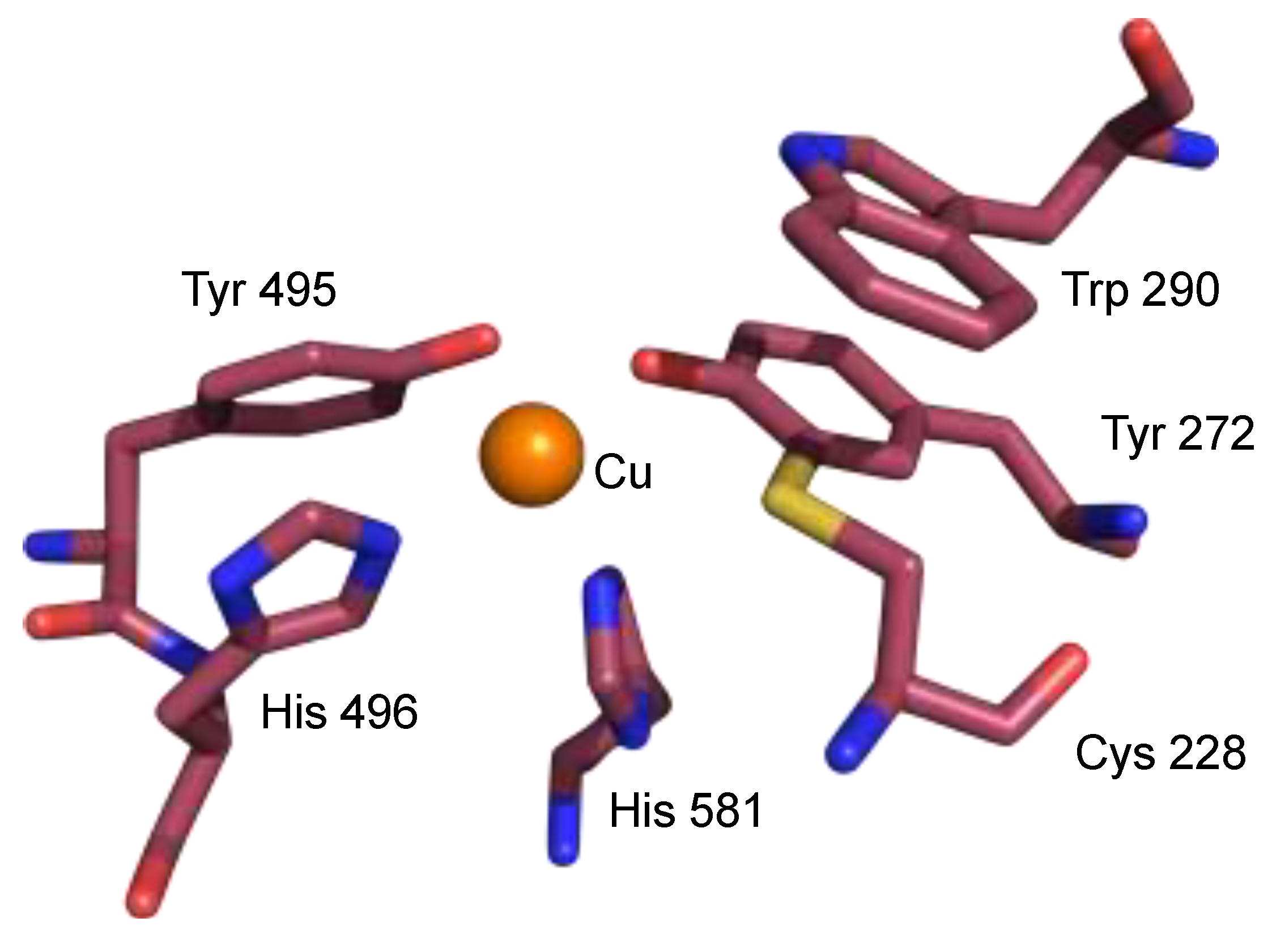
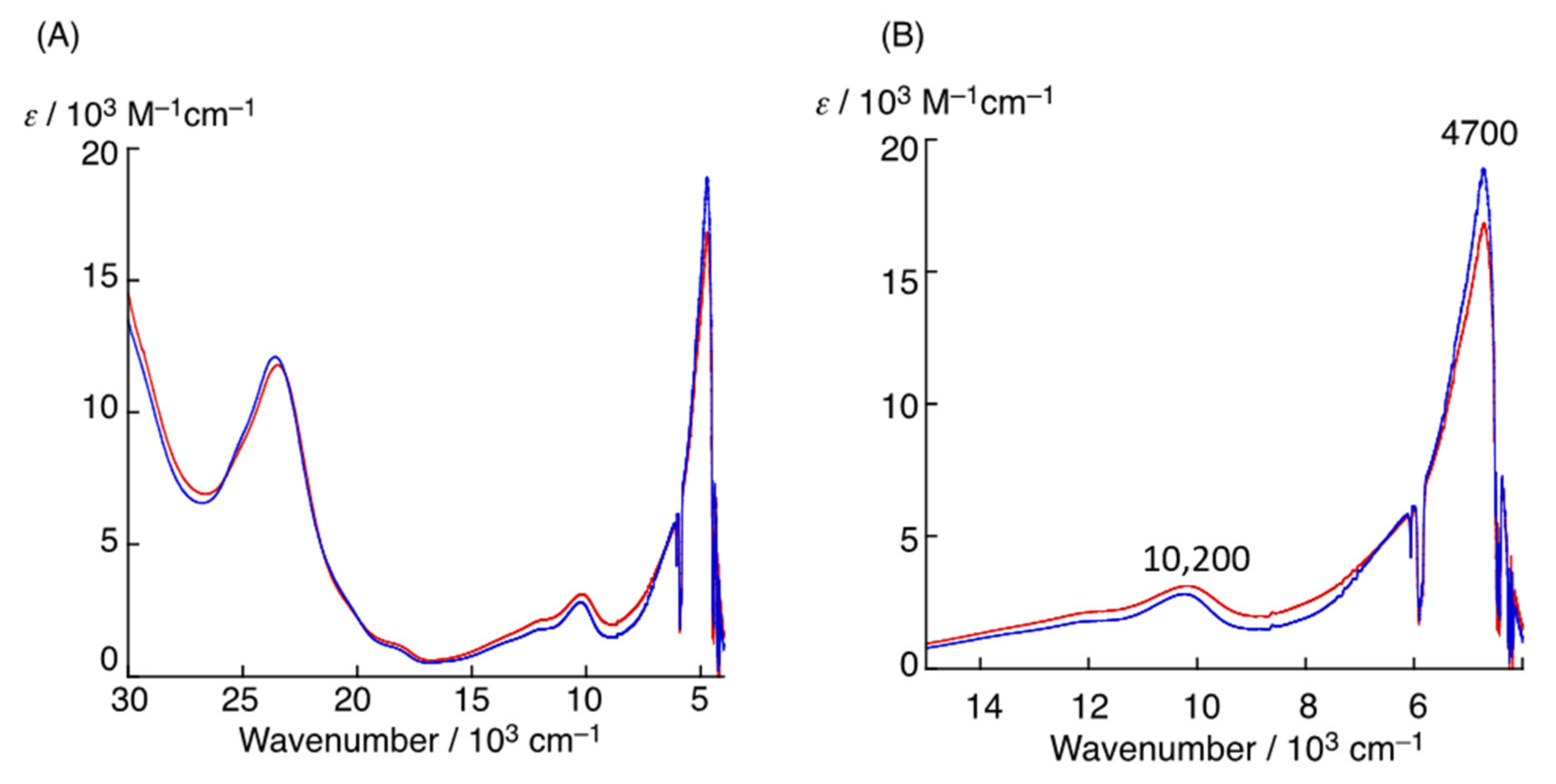
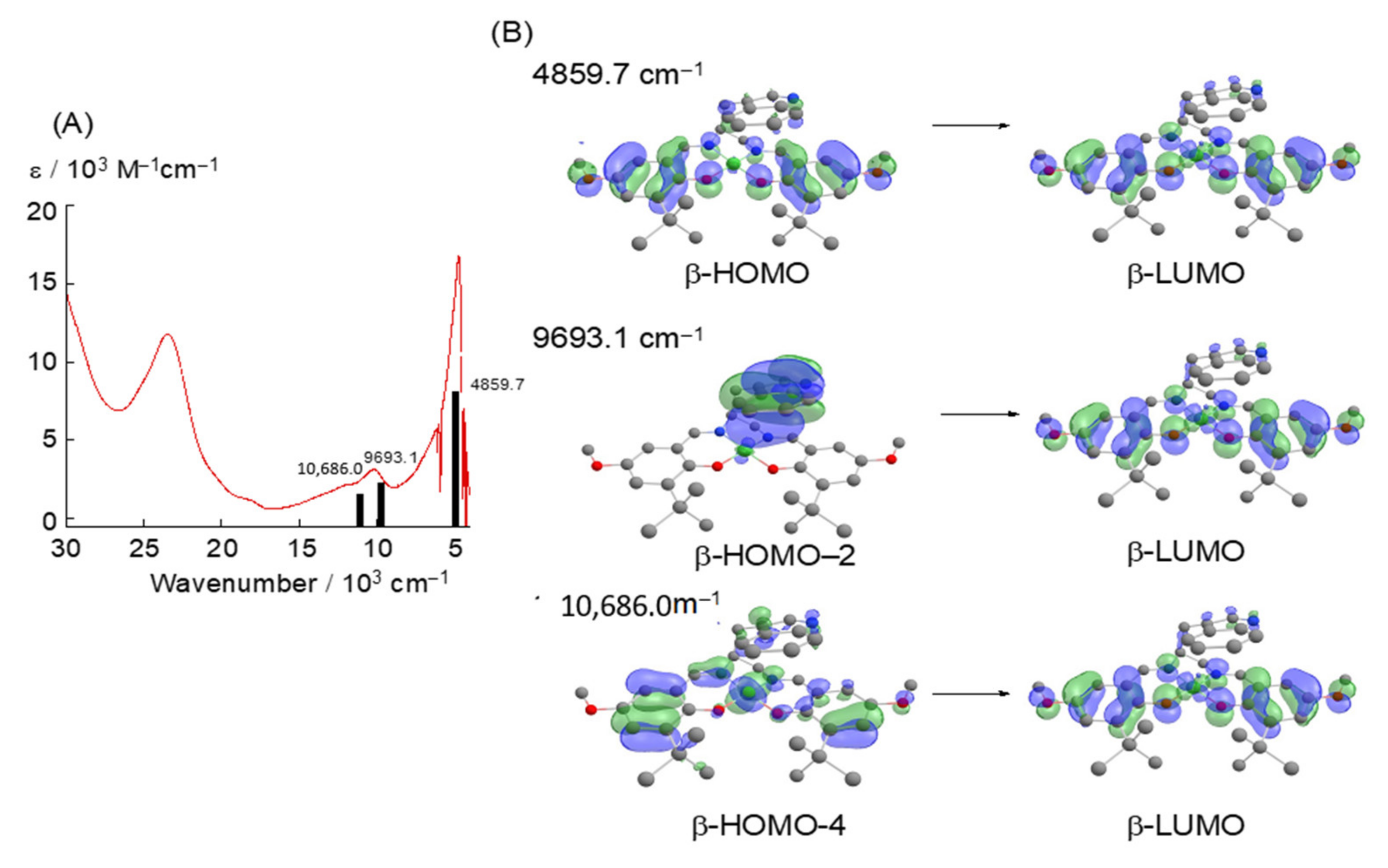
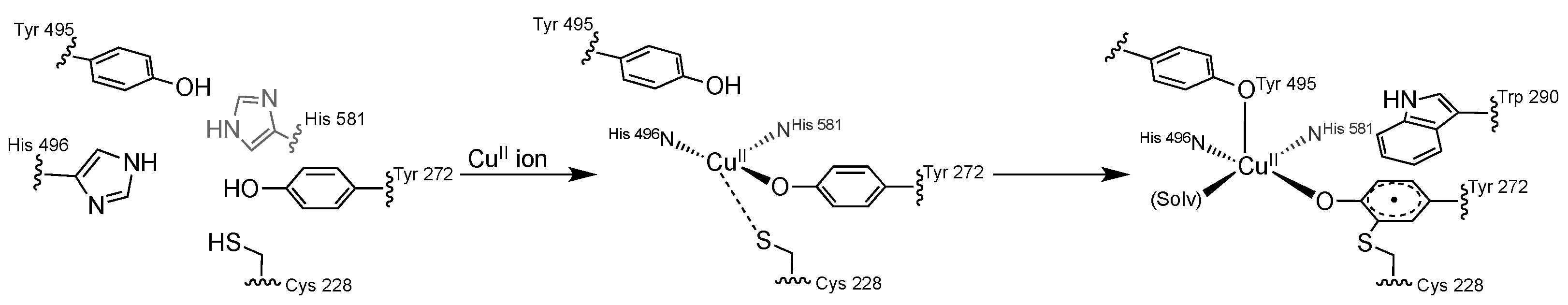
2. The π–π Stacking Interaction in the Active Site of GO
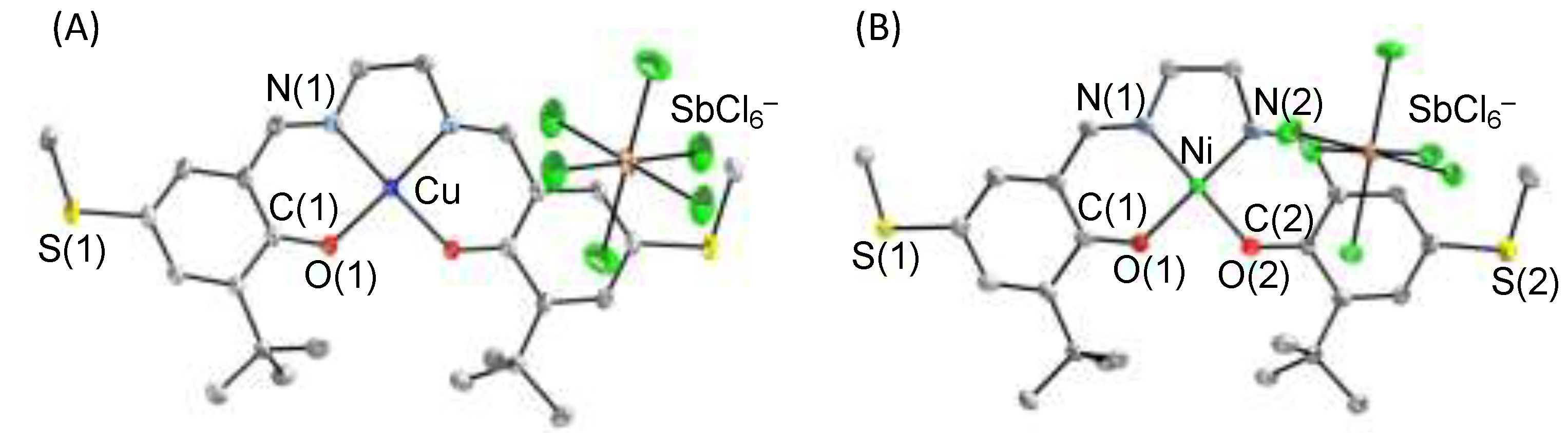
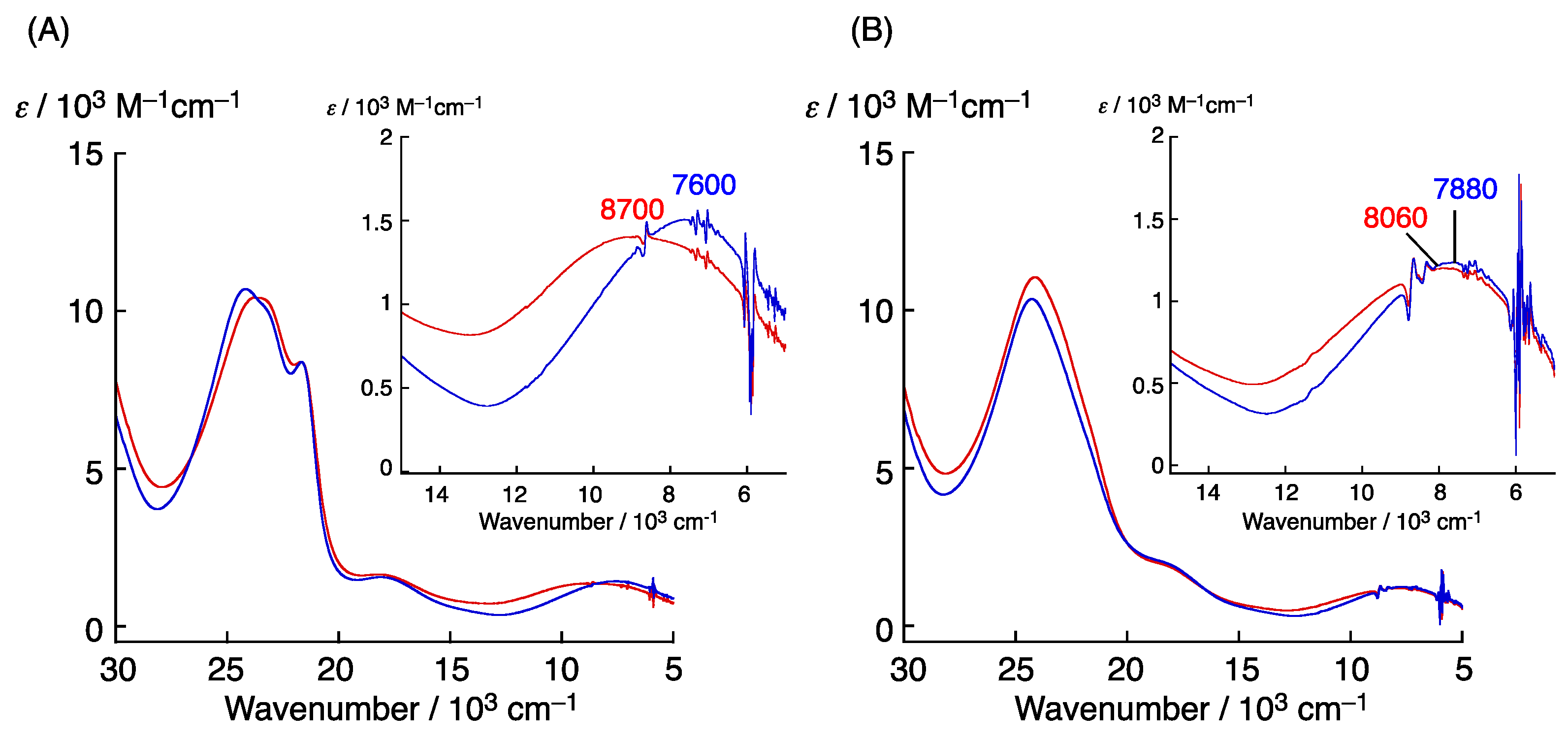
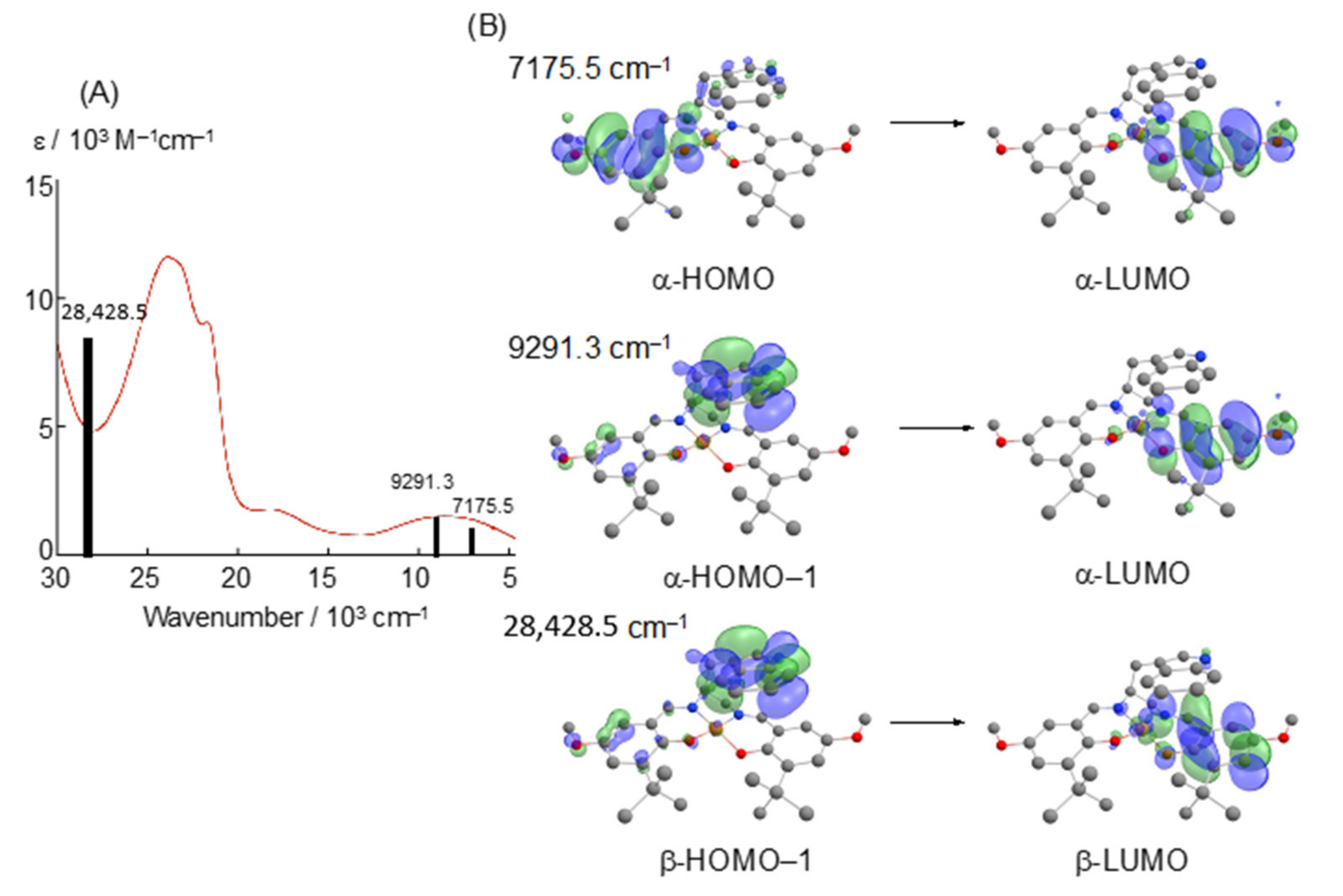
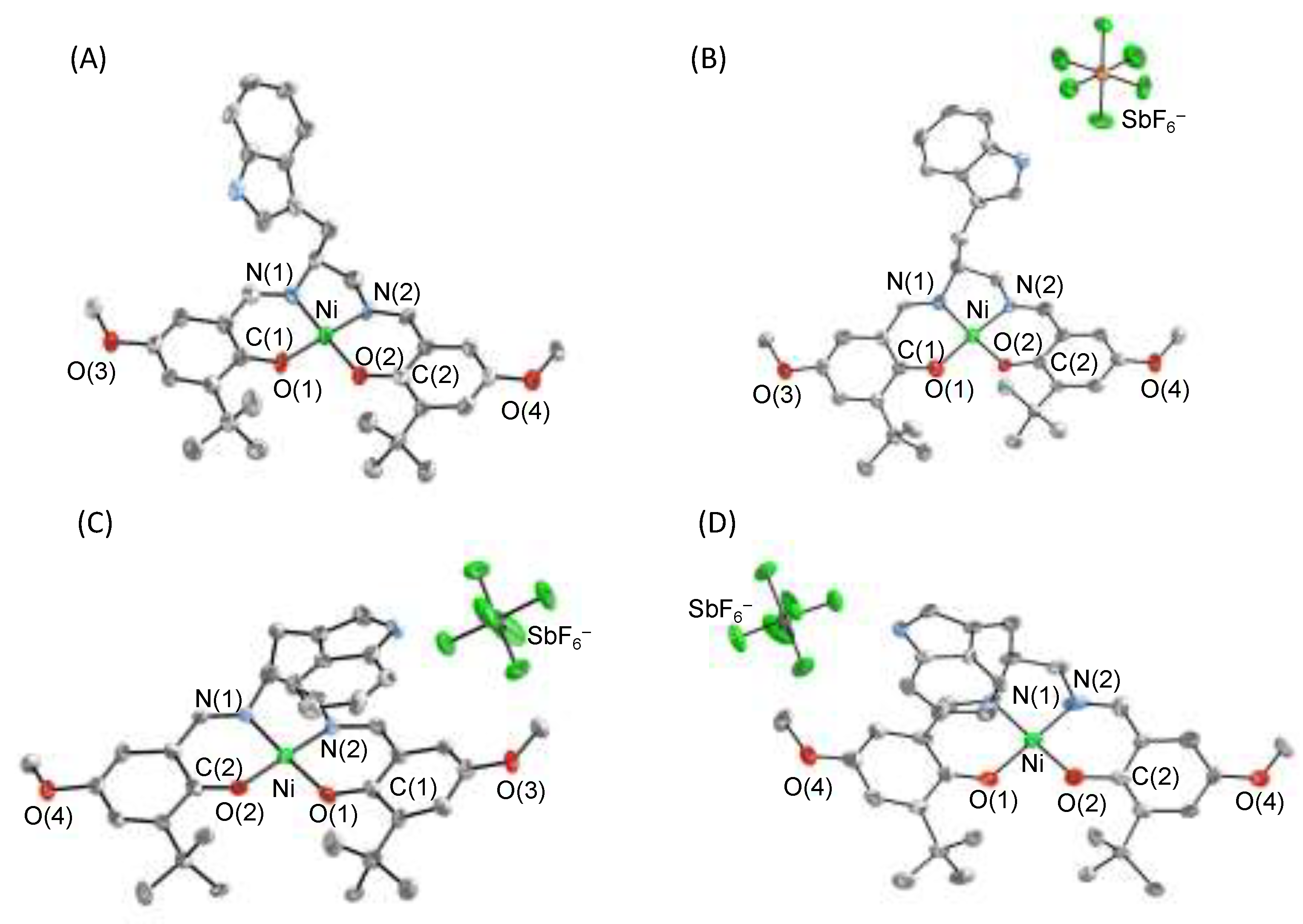
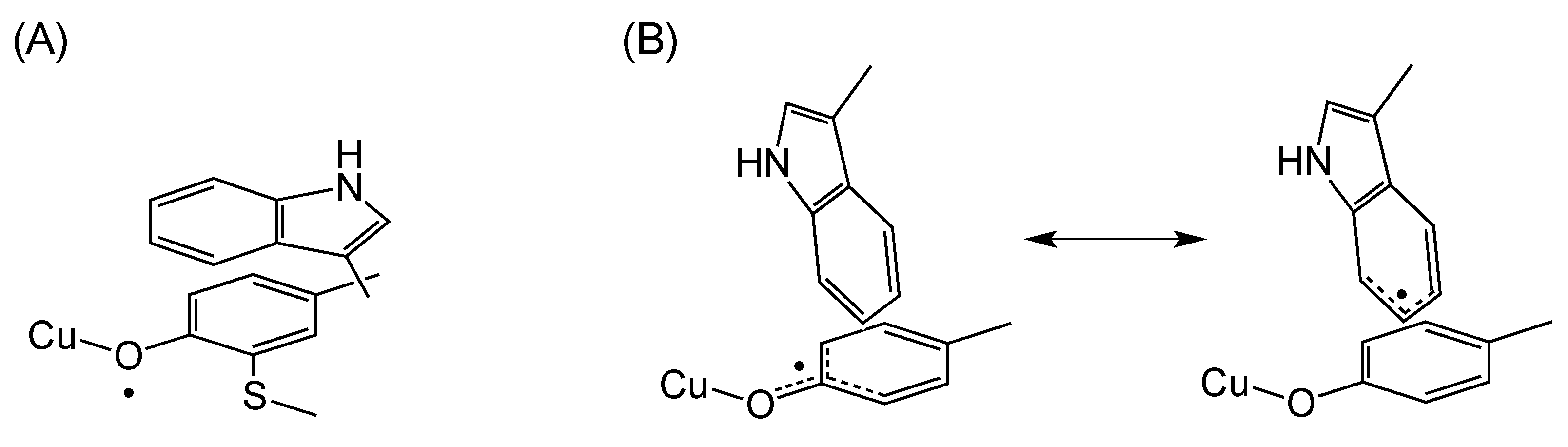
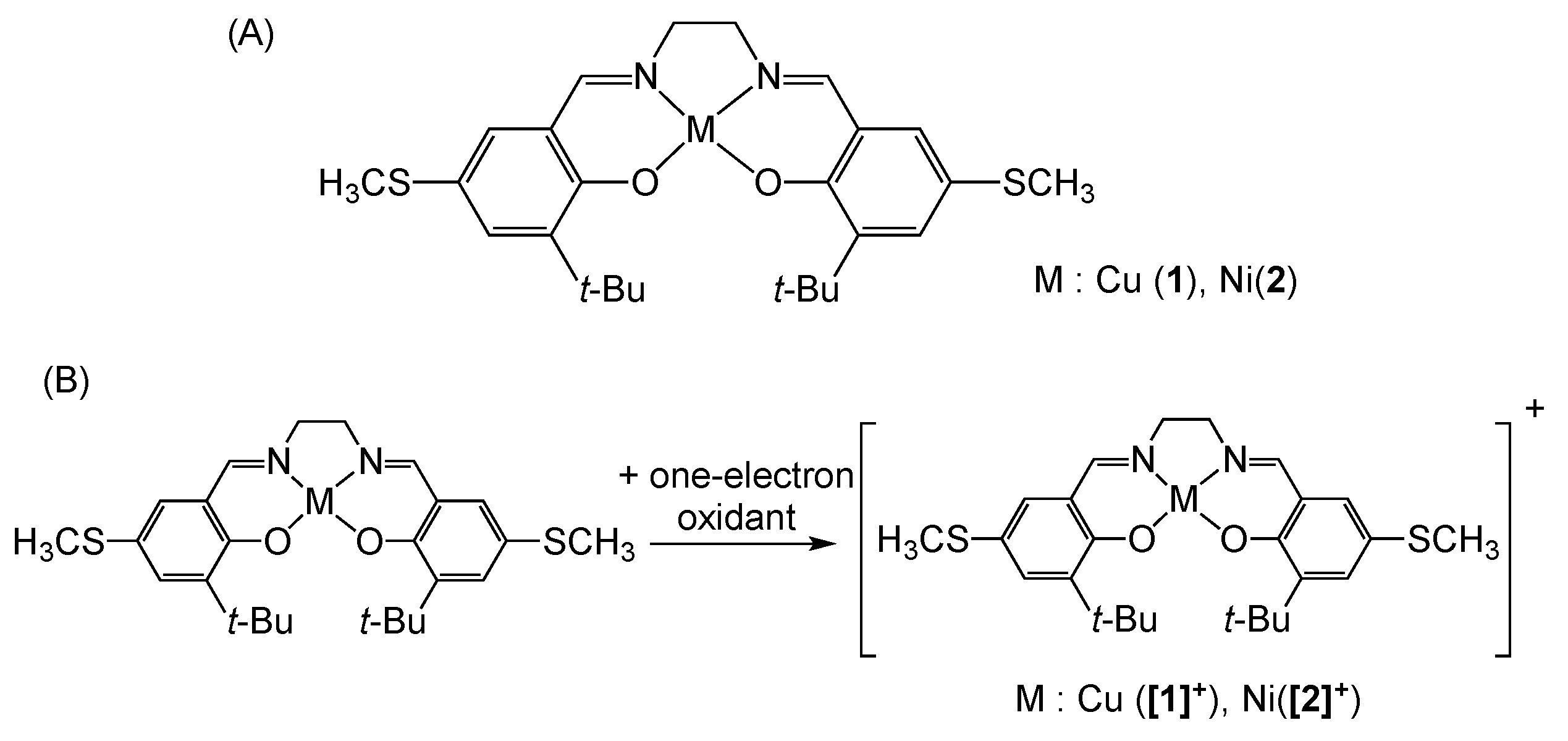
3. π–π Stacking Interaction of Methylthio-Phenoxyl Radical Metal Complexes
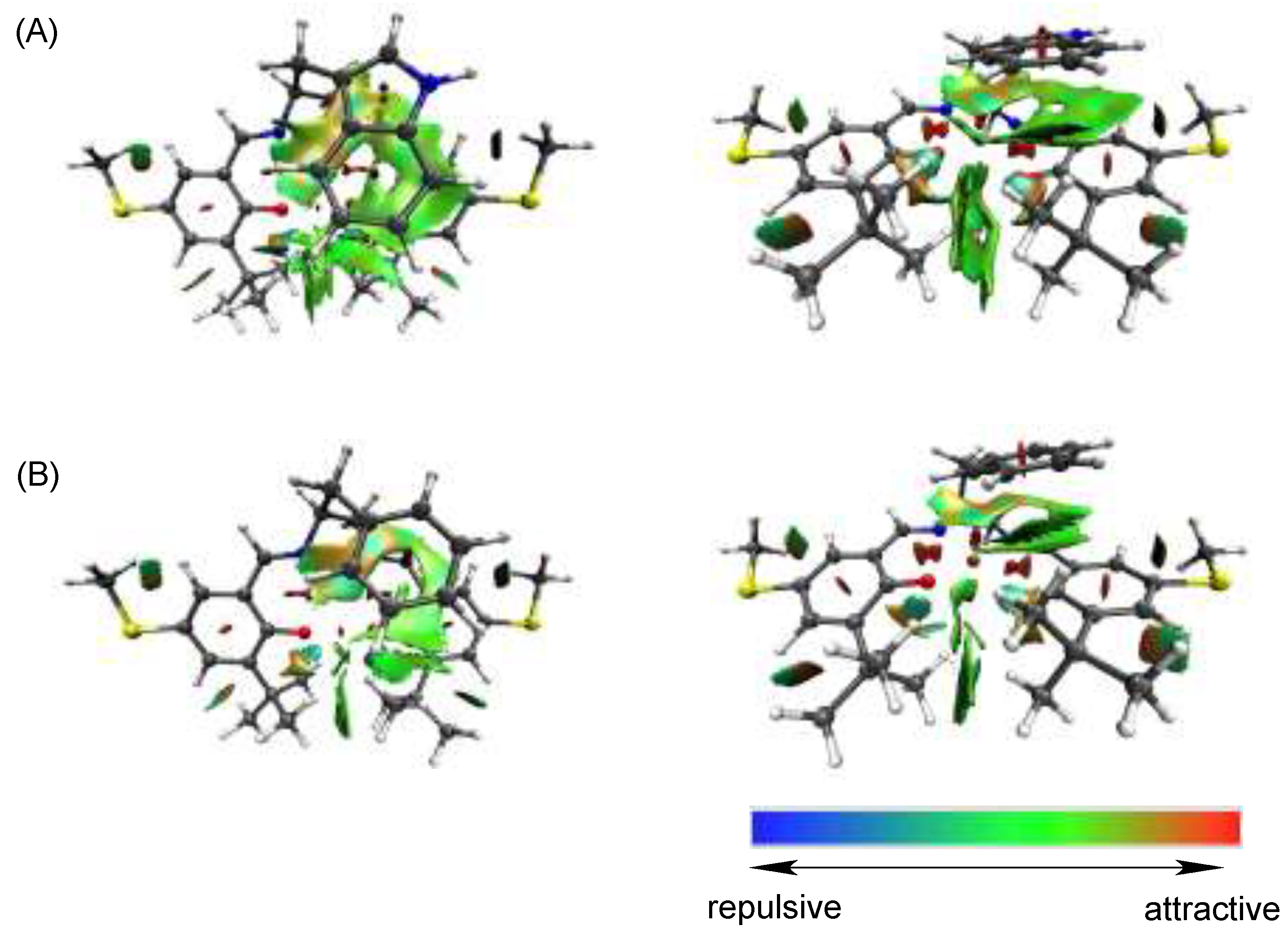
4. The π–π Stacking Interaction of Phenoxyl Radical with an Indole Ring
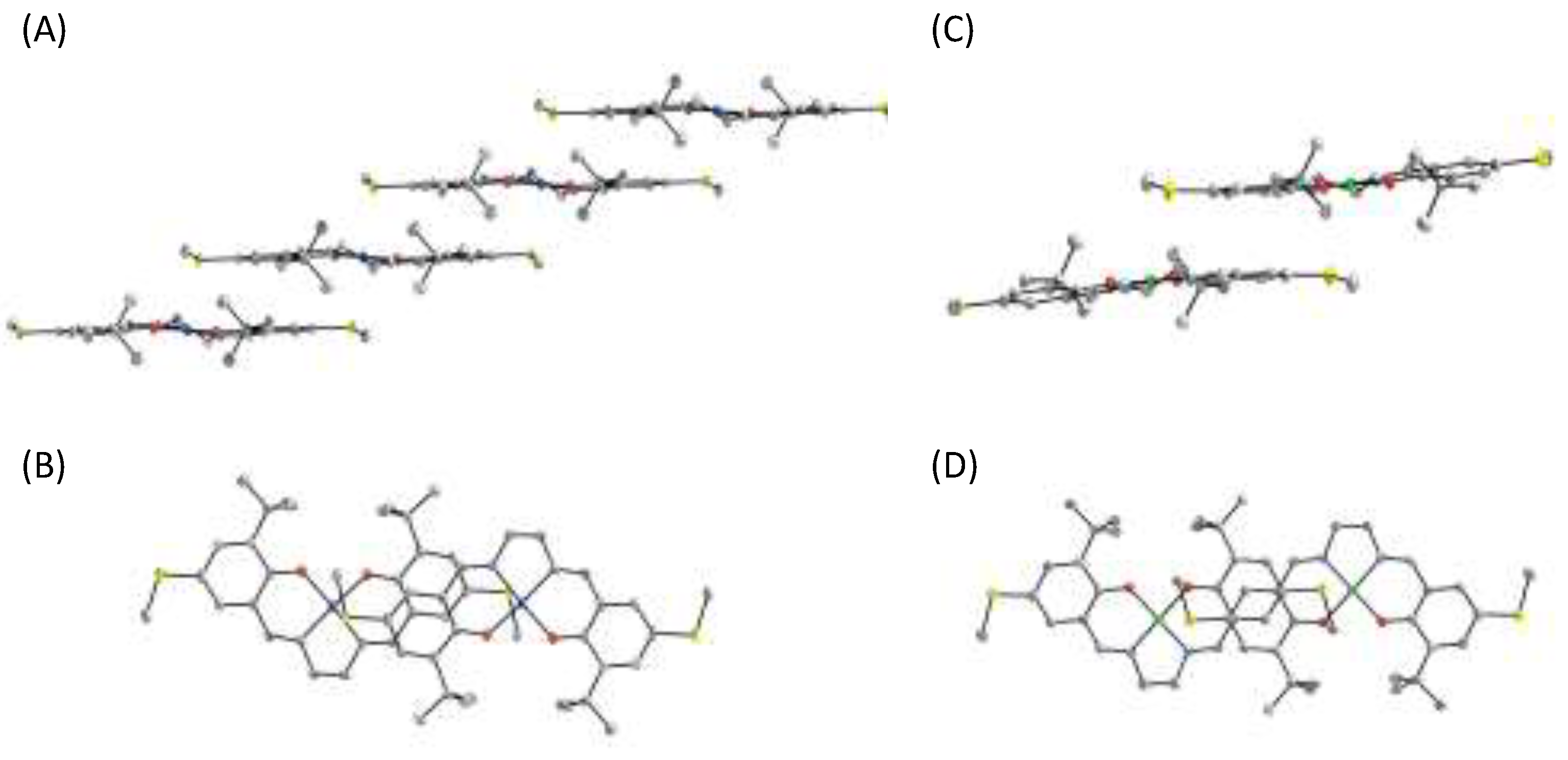
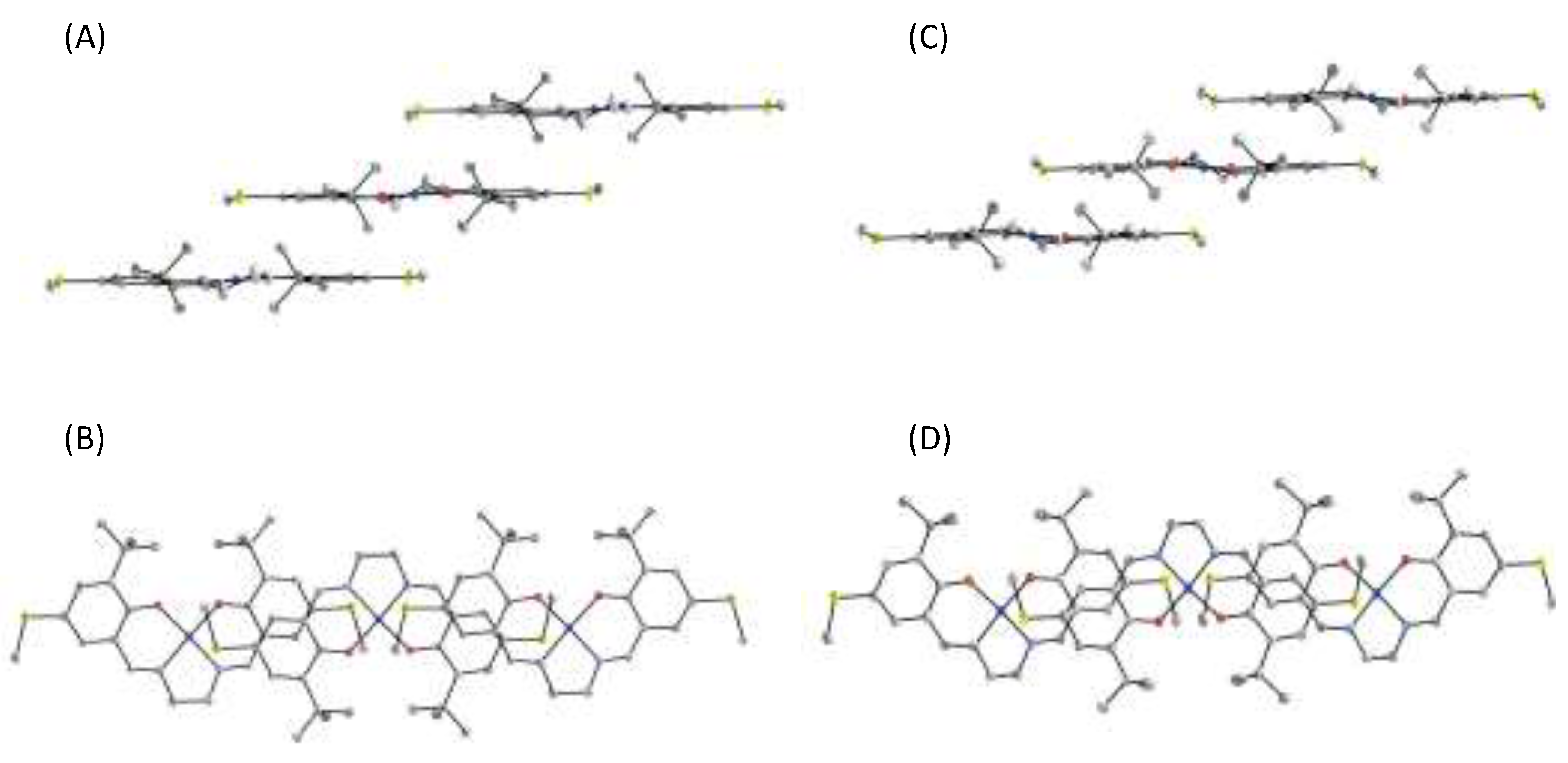
4.1. The π–π Stacking Interaction of Methoxyphenoxyl Radical with an Indole Ring
4.2. The π–π Stacking Interaction of Methylthiophenoxyl Radical with an Indole Ring
4.3. The Effect of the Electronic Structure of the Phenoxyl Radical on π–π Stacking Interaction with an Indole Ring
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamauchi, O. Non-covalent interactions in biocomplexes. In New-Generation Bioinorganic Complexes; Jastrzab, R., Tylkowski, B., Eds.; De Gruyter: Berlin, Germany, 2016; pp. 1–40. [Google Scholar]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell; Garland Publishing: New York, NY, USA, 1994; pp. 89–138. [Google Scholar]
- Hobza, P.; Müller-Dethlefs, K. Non-covalent Interactions: Theory and Experiment; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Scheiner, S.; Biswal, H.S.; Takahashi, O.; Li, Q.Z.; Li, H.B.; Kozelka, J.; Grabowski, S.J.; Del Bene, J.E.; Alkorta, I.; Elguero, J.; et al. Noncovalent Forces.; Scheiner, S., Ed.; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- Karshikoff, A. Noncovalent Interactions in Proteins; Imperial College Press: London, UK, 2006. [Google Scholar]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J.L.; Antonio, J.P.M.; Farias, G.D.V.; Santos, F.M.F.; Oliveira, R.; Cal, P.M.S.D.; et al. Noncovalent Interactions in the Synthesis and Design of New Compounds; Maharramov, A.M., Mahmudov, K.T., Kopylovich, M.N., Pombeiro, A.J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Burley, S.K.; Petsko, G.A. Stability of protein pharmaceuticals. Adv. Protein Chem. 1988, 39, 125–189. [Google Scholar] [PubMed]
- Guin, D.; Gruebele, M. Weak Chemical Interactions That Drive Protein Evolution: Crowding, Sticking, and Quinary Structure in Folding and Function. Chem. Rev. 2019, 119, 10691–10717. [Google Scholar] [CrossRef] [PubMed]
- Hopza, P.; Zahradnik, R. Weak Intermolecular Interactions in Chemistry and Biology; Elsevier Scientific Publishing: Amsterdam, The Netherlands, 1980. [Google Scholar]
- Dykstra, C.E. Electrostatic interaction potentials in molecular force fields. Chem. Rev. 1993, 93, 2339–2353. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Noncovalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Yamauchi, O.; Odani, A.; Takani, M. Metal-amino acid chemistry. Weak interaction and related functions of side chain groups. Dalton Trans. 2002, 3411–3421. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Aromatic-aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef]
- Mayer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Brown, C.J.; Toste, F.D.; Bergman, R.G.; Raymond, K.N. Supramolecular Catalysis in Metal−Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Dougherty, D.A. Cation–π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; da Silva, M.F.C.G.; Pombeiro, A.J.L.; Arimitsu, S.; Higashi, M.; Lillo, J.M.S.V.J.; Mansilla, J.; Sarazin, Y.; Carpentier, J.-F.; et al. Noncovalent Interactions in Catalysis; Mahmudov, K.T., Kopylovich, M.N., Guedes da Silva, M.F.C., Pombeiro, A.J.L., Eds.; Royal Society of Chemistry: Cambridge, UK, 2019. [Google Scholar]
- Dougherty, D.A. Cation−π Interactions Involving Aromatic Amino Acids. J. Nutrition. 2007, 137, 1504S–1508S. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation–π interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative quide. Proc. Natl. Acad. Sci. USA 1996, 93, 10566–10571. [Google Scholar]
- Dougherty, D.A. Cation–π interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar]
- Zarić, S.D. Metal ligand aromatic cation–π interactions. Eur. J. Inorg. Chem. 2003, 2197–2209. [Google Scholar] [CrossRef]
- Dougherty, D.A. The cation–π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef]
- Yamada, S. Cation−π Interactions in Organic Synthesis. Chem. Rev. 2018, 118, 11353–11432. [Google Scholar] [CrossRef]
- Collman, J.P.; Gagne, R.R.; Reed, C.A.; Robinson, W.T.; Rodley, G.A. Structure of an Iron(II) Dioxygen Complex; A Model for Oxygen Carrying Hemeproteins. Proc. Natl. Acad. Sci. USA 1974, 71, 1326–1329. [Google Scholar] [CrossRef]
- Borovic, A.S. Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res. 2005, 38, 54–61. [Google Scholar] [CrossRef]
- Shook, R.L.; Borovic, A.S. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem. 2010, 49, 3646–3660. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.W. Free radical catalysis by galactose oxidase. Chem. Rev. 2003, 103, 2347–2364. [Google Scholar] [CrossRef] [PubMed]
- Rokhsana, D.; Shepard, E.M.; Brown, D.E.; Dooley, D.M. Amine Oxidase and Galactose Oxidase. In Copper Oxygen Chemistry; Karlin, K.D., Itoh, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 53–106. [Google Scholar]
- Stubbe, J.; van der Donk, W.A. Protein Radicals in Enzyme Catalysis. Chem. Rev. 1998, 98, 705–762. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Whittaker, J.W. Catalytic Reaction Profile for Alcohol Oxidation by Galactose Oxidase. Biochemistry 2001, 40, 7140–7148. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Phillips, S.E.V.; Stevens, C.; Ogel, Z.B.; McPherson, M.J.; Keen, J.N.; Yadav, K.D.; Knowles, P.F. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Oshita, H.; Shimazaki, Y. Recent Advances in One-electron Oxidized CuII-Diphenoxide Complexes as Models of Galactose Oxidase: Importance of the Structural Flexibility in the Active Site. Chem. Eur. J. 2020, 26, 8324–8340. [Google Scholar] [CrossRef]
- Thomas, F. Metal Coordinated Phenoxyl Radicals. In Stable Radicals: Fundamentals and Applied Aspects of Odd Electron Compounds; Hicks, R.G., Ed.; John Wiley & Sons: Chichester, UK, 2010; pp. 281–316. [Google Scholar]
- Shimazaki, Y. Phenoxyl radical-metal complexes. In The Chemistry of Metal Phenolates; Zabicky, J., Ed.; John Wiley & Sons: Chichester, UK, 2014; Volume 1, pp. 593–668. [Google Scholar]
- Shimazaki, Y. Recent advances in the field of phenoxyl radical-metal complexes. In The Chemistry of Metal Phenolates Volume II; Zabicky, J., Ed.; John Wiley & Sons: Chichester, UK, 2016; pp. 269–294. [Google Scholar]
- Shimazaki, Y.; Takani, M.; Yamauchi, O. Metal complexes of amino acids and amino acid side chain groups. Structures and properties. Dalton Trans. 2009, 7854–7869. [Google Scholar] [CrossRef] [PubMed]
- Pierre, J.-L. One electron at a time oxidations and enzymatic paradigms: From metallic to non-metallic redox centers. Chem. Soc. Rev. 2000, 29, 251–257. [Google Scholar] [CrossRef]
- Jazdzewski, B.A.; Tolman, W.B. Understanding the Copper-Phenoxyl Radical Array in Galactose Oxidase: Contributions From Synthetic Modeling Studies. Coord. Chem. Rev. 2000, 200–202, 633–685. [Google Scholar] [CrossRef]
- Itoh, S.; Taki, M.; Fukuzumi, S. Active Site Models for Galactose Oxidase and Related Enzymes. Coord. Chem. Rev. 2000, 198, 3–20. [Google Scholar] [CrossRef]
- Thomas, F. Ten Years of a Biomimetic Approach to the Copper(II) Radical Site of Galactose Oxidase. Eur. J. Inorg. Chem. 2007, 17, 2379–2404. [Google Scholar] [CrossRef]
- Lyons, C.T.; Stack, T.D.P. Recent advances in phenoxyl radical complexes of salen-type ligands as mixed-valent galactose oxidase models. Coord. Chem. Rev. 2013, 257, 528–540. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Wieghardt, K. Phenoxyl radical complexes. Prog. Inorg. Chem. 2001, 50, 151–216. [Google Scholar]
- Dyrkacz, G.R.; Libby, R.D.; Hamilton, G.A. Trivalent copper as a probable intermediate in the reaction catalyzed by galactose oxidase. J. Am. Chem. Soc. 1976, 98, 626–628. [Google Scholar] [CrossRef]
- Hamilton, G.A.; Adolf, P.K.; de Jersey, J.; DuBois, G.C.; Dyrkacz, G.R.; Libby, R.D. Trivalent copper, superoxide, and galactose oxidase. J. Am. Chem. Soc. 1978, 100, 1899–1912. [Google Scholar] [CrossRef]
- Clark, K.; Penner-Hahn, J.E.; Whittaker, M.M.; Whittaker, J.W. Oxidation-state assignments for galactose oxidase complexes from x-ray absorption spectroscopy. Evidence for copper(II) in the active enzyme. J. Am. Chem. Soc. 1990, 112, 6433–6434. [Google Scholar] [CrossRef]
- McGlashen, M.L.; Eads, D.D.; Spiro, T.G.; Whittaker, J.W. Resonance Raman Spectroscopy of Galactose Oxidase: A New Interpretation Based on Model Compound Free Radical Spectra. J. Phys. Chem. 1995, 99, 4918–4922. [Google Scholar] [CrossRef]
- Storr, T.; Verma, P.; Pratt, R.C.; Wasinger, E.C.; Shimazaki, Y.; Stack, T.D.P. Defining the Electronic and Geometric Structure of One-Electron Oxidized Copper−Bis-phenoxide Complexes. J. Am. Chem. Soc. 2008, 130, 15448–15459. [Google Scholar] [CrossRef]
- Asami, K.; Tsukidate, K.; Iwatsuki, S.; Tani, F.; Karasawa, S.; Chiang, L.; Storr, T.; Thomas, F.; Shimazaki, Y. New Insights into the Electronic Structure and Reactivity of One-Electron Oxidized Copper(II)-(Disalicylidene)diamine Complexes. Inorg. Chem. 2012, 51, 12450–12461. [Google Scholar] [CrossRef] [PubMed]
- Asami, K.; Takashina, A.; Kobayashi, M.; Iwatsuki, S.; Yajima, T.; Kochem, A.; van Gastel, M.; Tani, F.; Kohzuma, T.; Thomas, F.; et al. Characterization of one-electron oxidized copper(ii)-salophen-type complexes; effects of electronic and geometrical structures on reactivities. Dalton Trans. 2014, 43, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Yajima, T.; Yamauchi, O. Properties of the indole ring in metal complexes. A comparison with the phenol ring. J. Inorg. Biochem. 2015, 148, 105–115. [Google Scholar] [CrossRef]
- Avigad, G.; Amaral, D.; Asensio, C.; Horecker, B.L. The D-galactose oxidase of Polyporus circinatus. J. Biol. Chem. 1962, 237, 2736–2743. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Whittaker, J.W. Cu(I)-dependent Biogenesis of the Galactose Oxidase Redox Cofactor. J. Biol. Chem. 2003, 278, 22090–22101. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.J.; Stevens, C.; Wilmot, C.M.; Knowles, P.F.; Phillips, S.E.V.; McPherson, M.J. Preliminary studies of two active site mutants of galactose oxidase. Biochem. Soc. Trans. 1993, 21, 319S. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.S.; Tyler, E.M.; Akyumani, N.; Kurtis, C.R.; Spooner, R.K.; Deacon, S.E.; Tamber, S.; Firbank, S.J.; Mahmoud, K.; Knowles, P.F.; et al. The stacking tryptophan of galactose oxidase: A second coordination sphere residue that has profound effects on tyrosyl radical behavior and enzyme catalysis. Biochemistry 2007, 46, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Firbank, S.J.; Rogers, M.S.; Wilmot, C.M.; Dooley, D.M.; Halcrow, M.A.; Knowles, P.F.; McPherson, M.J.; Phillips, S.E.V. Crystal structure of the precursor of galactose oxidase: An unusual self-processing enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 12932–12937. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.S.; Hurtado-Guerrero, R.; Firbank, S.J.; Halcrow, M.A.; Dooley, D.M.; Phillips, S.E.V.; Knowles, P.F.; McPherson, M.J. Cross-Link Formation of the Cysteine 228−Tyrosine 272 Catalytic Cofactor of Galactose Oxidase Does Not Require Dioxygen. Biochemistry 2008, 47, 10428–10439. [Google Scholar] [CrossRef]
- Chaplin, A.K.; Bernini, C.; Sinicropi, A.; Basosi, R.; Worrall, J.A.R.; Svistunenko, D.A. Tyrosine or Tryptophan? Modifying a Metalloradical Catalytic Site by Removal of the Cys–Tyr Cross-Link in the Galactose 6-Oxidase Homologue GlxA. Angew. Chem. Int. Ed. 2017, 56, 6502–6506. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An Unusual Pi-Stacking Interaction. Chem. Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y. Recent Advances in X-Ray Structures of Metal-Phenoxyl Radical Complexes. Adv. Mater. Phys. Chem. 2013, 3, 60–71. [Google Scholar] [CrossRef]
- Takeyama, T.; Suzuki, T.; Kikuchi, M.; Kobayashi, M.; Oshita, H.; Kawashima, K.; Mori, S.; Abe, H.; Hoshino, N.; Iwatsuki, S.; et al. Solid State Characterization of One- and Two-Electron Oxidized CuII-salen Complexes with para-Substituents: Geometric Structure-Magnetic Property Relationship. Eur. J. Inorg. Chem. 2021, 2021, 4133–4145. [Google Scholar] [CrossRef]
- Oshita, H.; Kikuchi, M.; Mieda, K.; Ogura, T.; Yoshimura, T.; Tani, F.; Yajima, T.; Abe, H.; Mori, S.; Shimazaki, Y. Characterization of Group 10-Metal-p-Substituted Phenoxyl Radical Complexes with Schiff Base Ligands. Chemistry Select 2017, 2, 10221–10231. [Google Scholar] [CrossRef]
- Verma, P.; Pratt, R.C.; Storr, T.; Wasinger, E.C.; Stack, T.D.P. Sulfanyl stabilization of copper-bonded phenoxyls in model complexes and galactose oxidase. Proc. Natl. Acad. Sci. USA 2011, 108, 18600–18605. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.C.; Lyons, C.T.; Wasinger, E.C.; Stack, T.D.P. Electrochemical and Spectroscopic Effects of Mixed Substituents in Bis(phenolate)–Copper(II) Galactose Oxidase Model Complexes. J. Am. Chem. Soc. 2012, 134, 7367–7377. [Google Scholar] [CrossRef][Green Version]
- Neel, A.J.; Hilton, M.J.; Sigman, M.S.; Toste, F.D. Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.B.; Day, P. Mixed Valence Chemistry-A Survey and Classification. Adv. Inorg. Chem. Radiochem. 1968, 10, 247–422. [Google Scholar]
- Creutz, C.; Taube, H. Direct approach to measuring the Franck-Condon barrier to electron transfer between metal ions. J. Am. Chem. Soc. 1969, 91, 3988–3989. [Google Scholar] [CrossRef]
- Hush, N.S. Distance Dependence of Electron Transfer Rates. Coord. Chem. Rev. 1985, 64, 135–157. [Google Scholar] [CrossRef]
- Kanso, H.; Clarke, R.M.; Kochem, A.; Arora, H.; Philouze, C.; Jarjayes, O.; Storr, T.; Thomas, F. Effect of Distortions on the Geometric and Electronic Structures of One-Electron Oxidized Vanadium(IV), Copper(II), and Cobalt(II)/(III) Salen Complexes. Inorg. Chem. 2020, 59, 5133–5148. [Google Scholar] [CrossRef]
- Hua, C.; Doheny, P.W.; Ding, B.; Chan, B.; Yu, M.; Kepert, C.J.; D’Alessandro, D.M. Through-Space Intervalence Charge Transfer as a Mechanism for Charge Delocalization in Metal–Organic Frameworks. J. Am. Chem. Soc. 2018, 140, 6622–6630. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Stack, T.D.P.; Storr, T. Detailed Evaluation of the Geometric and Electronic Structures of One-electron Oxidized Group 10 (Ni, Pd, and Pt) Metal(II)-(Disalicylidene)diamine Complexes. Inorg. Chem. 2009, 48, 8383–8392. [Google Scholar] [CrossRef]
- Müller, J.; Weyhermüller, T.; Bill, E.; Hildebrandt, P.; Moussa, L.O.; Glaser, T.; Wieghardt, K. Why Does the Active Form of Galactose Oxidase Possess a Diamagnetic Ground State? Angew. Chem. Int. Ed. 1998, 37, 616–619. [Google Scholar] [CrossRef]
- Bill, E.; Müller, J.; Weyhermüller, T.; Wieghardt, K. Intramolecular Spin Interactions in Bis(phenoxyl)metal Complexes of Zinc(II) and Copper(II). Inorg. Chem. 1999, 38, 5795–5802. [Google Scholar] [CrossRef]
- Oshita, H.; Suzuki, T.; Kawashima, K.; Abe, H.; Tani, F.; Mori, S.; Yajima, T.; Shimazaki, Y. π–π Stacking Interaction in an Oxidized CuII–Salen Complex with a Side-Chain Indole Ring: An Approach to the Function of the Tryptophan in the Active Site of Galactose Oxidase. Chem. Eur. J. 2019, 25, 7649–7658. [Google Scholar] [CrossRef] [PubMed]
- Oshita, H.; Yoshimura, T.; Mori, S.; Tani, F.; Shimazaki, Y.; Yamauchi, O. Characterization of the one-electron oxidized Cu(II)-salen complexes with a side chain aromatic ring: The effect of the indole ring on the Cu(II)-phenoxyl radical species. J. Biol. Inorg. Chem. 2018, 23, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Oshita, H.; Suzuki, T.; Kawashima, K.; Abe, H.; Tani, F.; Mori, S.; Yajima, T.; Shimazaki, Y. The effect of π–π stacking interaction of the indole ring with the coordinated phenoxyl radical in a nickel(II)-salen type complex. Comparison with the corresponding Cu(II) complex. Dalton Trans. 2019, 48, 12060–12069. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Yajima, T.; Takani, M.; Yamauchi, O. Metal complexes involving indole rings: Structures and effects of metal–indole interactions. Coord. Chem. Rev. 2009, 253, 479–492. [Google Scholar] [CrossRef]
- Sugimori, T.; Masuda, H.; Ohata, N.; Koiwai, K.; Odani, A.; Yamauchi, O. Structural Dependence of Aromatic Ring Stacking and Related Weak Interactions in Ternary Amino Acid−Copper(II) Complexes and Its Biological Implication. Inorg. Chem. 1997, 36, 576–583. [Google Scholar] [CrossRef]
- Yajima, T.; Shimazaki, Y.; Ishigami, N.; Odani, A.; Yamauchi, O. Conformational preference of the side chain aromatic ring in Cu(II) and Pd(II) complexes of 2N1O-donor ligands. Inorg. Chem. Acta. 2002, 337, 193–202. [Google Scholar] [CrossRef]
- Yajima, T.; Takamido, R.; Shimazaki, Y.; Odani, A.; Nakabayashi, Y.; Yamauchi, O. π–π Stacking assisted binding of aromatic amino acids by copper(II)–aromatic diimine complexes. Effects of ring substituents on ternary complex stability. Dalton Trans. 2007, 3, 299–307. [Google Scholar] [CrossRef][Green Version]
- Taborosi, A.; Yamaguchi, T.; Odani, A.; Yamauchi, O.; Kohzuma, T. The Role for the Weak Interaction on the Stabilization of Copper-containing Complex: DFT Investigation of Noncovalent Interactions in ternary-Cu(II) (DA)(AA) Complexes (DA=diamine and AA=amino acids) as a Model of Metalloprotein. Bull. Chem. Soc. Jpn. 2019, 92, 1874–1882. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The Nature of π–π Interaction. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Yamauchi, O.; Odani, A.; Hirota, S. Metal Ion-Assisted Weak Interactions Involving Biological Molecules. From Small Complexes to Metalloproteins. Bull. Chem. Soc. Jpn. 2001, 74, 1525–1545. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-Grarcía, J.; Cohen, A.J.; Yang, W. Revealing Non-Covalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Grarcía, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory. Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Yajima, T.; Tani, F.; Karasawa, S.; Fukui, K.; Naruta, Y.; Yamauchi, O. Syntheses and Electronic Structures of One-Electron-Oxidized Group 10 Metal(II)-(Disalicylidene)diamine Complexes (Metal = Ni, Pd, Pt). J. Am. Chem. Soc. 2007, 129, 2559–2568. [Google Scholar] [CrossRef]
- Thomas, F. Ligand-centred oxidative chemistry in sterically hindered salen complexes: An interesting case with nickel. Dalton Trans. 2016, 45, 10866–10877. [Google Scholar] [CrossRef]
- Storr, T.; Wasinger, E.C.; Pratt, R.C.; Stack, T.D.P. The Geometric and Electronic Structure of a One-Electron-Oxidized Nickel(II) Bis(salicylidene)diamine Complex. Angew. Chem., Int. Ed. 2007, 46, 5198–5201. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Arai, N.; Dunn, T.J.; Yajima, T.; Tani, F.; Ramogida, C.F.; Storr, T. Influence of the chelate effect on the electronic structure of one-electron oxidized group 10 metal(ii)-(disalicylidene)diamine complexes. Dalton Trans. 2011, 40, 2469–2479. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Tani, F.; Fukui, K.; Naruta, Y.; Yamauchi, O. One-Electron Oxidized Nickel(II)-(Disalicylidene)diamine Complex: Temperature-Dependent Tautomerism between Ni(III)-Phenolate and Ni(II)-Phenoxyl Radical States. J. Am. Chem. Soc. 2003, 125, 10512–10513. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Yamaguchi, T.; Masaoka, S.; Tani, F.; Kohzuma, T.; Chiang, L.; Mieda, K.; Ogura, T.; Szilagyi, R.K.; Shimazaki, Y. Influence of Ligand Flexibility on the Electronic Structure of Oxidized NiIII-Phenoxide Complexes. Inorg. Chem. 2014, 53, 10195–10202. [Google Scholar] [CrossRef] [PubMed]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oshita, H.; Shimazaki, Y. π–π Stacking Interaction of Metal Phenoxyl Radical Complexes. Molecules 2022, 27, 1135. https://doi.org/10.3390/molecules27031135
Oshita H, Shimazaki Y. π–π Stacking Interaction of Metal Phenoxyl Radical Complexes. Molecules. 2022; 27(3):1135. https://doi.org/10.3390/molecules27031135
Chicago/Turabian StyleOshita, Hiromi, and Yuichi Shimazaki. 2022. "π–π Stacking Interaction of Metal Phenoxyl Radical Complexes" Molecules 27, no. 3: 1135. https://doi.org/10.3390/molecules27031135
APA StyleOshita, H., & Shimazaki, Y. (2022). π–π Stacking Interaction of Metal Phenoxyl Radical Complexes. Molecules, 27(3), 1135. https://doi.org/10.3390/molecules27031135