
Sitagliptin Is More Effective Than Gliclazide in Preventing Pro-Fibrotic and Pro-Inflammatory Changes in a Rodent Model of Diet-Induced Non-Alcoholic Fatty Liver Disease


 ,
,
Abstract
:1. Introduction
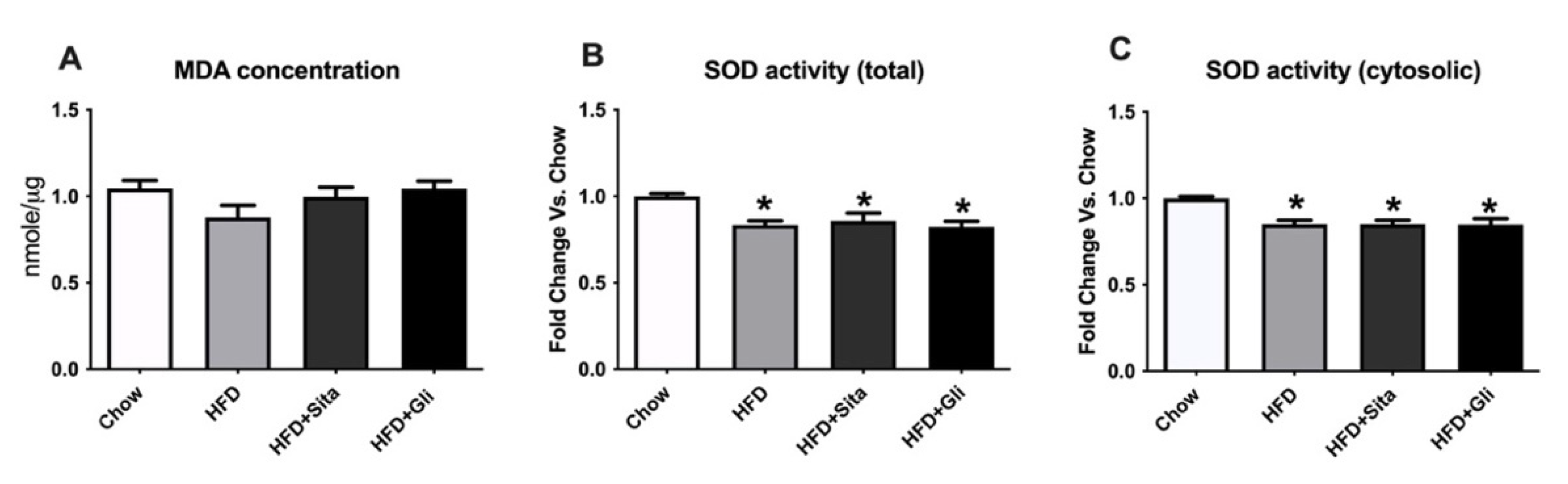
2. Results
2.1. Animal Profiles and Metabolic Measures
2.2. Sitagliptin Improves Steatosis and Inflammation
2.3. Sitagliptin Normalizes Fibrotic Markers
2.4. Exploration of Possible Underlying Mechanisms for Liver Protection by Sitagliptin
3. Discussion
4. Materials and Methods
4.1. Animal and Experimental Design
4.2. Blood Sampling and Assays
4.3. Quantitative Real-Time Polymerase Chain Reaction
4.4. Tissue Preparation and Histological Studies, including Staining Scoring
4.5. Western Immunoblot
4.6. Oxidative Stress Measurement
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Younossi, Z.M. Non-alcoholic fatty liver disease-A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver. Int. 2017, 37 (Suppl. 1), 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, B. Fatty liver disease: The liver labyrinth. Nature 2014, 516, S8–S9. [Google Scholar] [CrossRef]
- Williams, K.H.; Shackel, N.A.; Gorrell, M.D.; McLennan, S.V.; Twigg, S.M. Diabetes and nonalcoholic Fatty liver disease: A pathogenic duo. Endocr. Rev. 2013, 34, 84–129. [Google Scholar] [CrossRef] [PubMed]
- Thuluvath, P.J.; Hanish, S.; Savva, Y. Waiting List Mortality and Transplant Rates for NASH Cirrhosis When Compared With Cryptogenic, Alcoholic, or AIH Cirrhosis. Transplantation 2019, 103, 113–121. [Google Scholar] [CrossRef]
- Milic, S.; Lulic, D.; Stimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. WJG 2014, 20, 9330–9337. [Google Scholar] [PubMed]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef]
- Geelhoed-Duijvestijn, P.H. Incretins: A new treatment option for type 2 diabetes? Neth. J. Med. 2007, 65, 60–64. [Google Scholar]
- Ahren, B.; Landin-Olsson, M.; Jansson, P.A.; Svensson, M.; Holmes, D.; Schweizer, A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 2078–2084. [Google Scholar] [CrossRef] [Green Version]
- Mentzel, S.; Dijkman, H.B.; Van Son, J.P.; Koene, R.A.; Assmann, K.J. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1996, 44, 445–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, Y.H.; Korkusuz, P.; Simsek, H.; Gokcan, H.; Gedikoglu, G.; Pinar, A.; Hascelik, G.; Asan, E.; Hamaloglu, E.; Tatar, G. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann. Hepatol. 2007, 6, 242–250. [Google Scholar] [CrossRef]
- Barchetta, I.; Ceccarelli, V.; Cimini, F.A.; Barone, E.; Sentinelli, F.; Coluzzi, M.; Chiappetta, C.; Bertoccini, L.; Tramutola, A.; Labbadia, G.; et al. Circulating dipeptidyl peptidase-4 is independently associated with the presence and severity of NAFLD/NASH in individuals with and without obesity and metabolic disease. J. Endocrinol. Investig. 2021, 44, 979–988. [Google Scholar] [CrossRef]
- Akaslan, S.B.; Degertekin, C.K.; Yilmaz, G.; Cakir, N.; Arslan, M.; Toruner, F.B. Effects of sitagliptin on nonalcoholic fatty liver disease in diet-induced obese rats. Metab. Syndr. Relat. Disord. 2013, 11, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Ghosh, J.; Mustafa, G.; Kamal, M.; Ahmad, N. Effect of sitagliptin on hepatic histological activity and fibrosis of nonalcoholic steatohepatitis patients: A 1-year randomized control trial. Hepat Med. 2018, 10, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onoyama, T.; Koda, M.; Okamoto, T.; Kishina, M.; Matono, T.; Sugihara, T.; Murawaki, Y. Therapeutic effects of the dipeptidyl peptidase-IV inhibitor, sitagliptin, on non-alcoholic steatohepatitis in FLS-ob/ob male mice. Mol. Med. Rep. 2015, 12, 6895–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Gordon, S.; Hamann, J.; Lin, H.H.; Stacey, M. F4/80 and the related adhesion-GPCRs. Eur. J. Immunol. 2011, 41, 2472–2476. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease. Liver. Res. 2018, 2, 112–119. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Pipatpiboon, N.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 2012, 153, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. Dipeptidyl peptidase-4 inhibitors: Clinical data and clinical implications. Diabetes Care 2007, 30, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A. Treating diabetes today with gliclazide MR: A matter of numbers. Diabetes Obes. Metab. 2012, 14 (Suppl. 1), 14–19. [Google Scholar] [CrossRef] [PubMed]
- Briand, F.; Thieblemont, Q.; Burcelin, R.; Sulpice, T. Sitagliptin promotes macrophage-to-faeces reverse cholesterol transport through reduced intestinal cholesterol absorption in obese insulin resistant CETP-apoB100 transgenic mice. Diabetes Obes. Metab. 2012, 14, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Sallas, W.M.; He, Y.L.; Watson, C.; Ligueros-Saylan, M.; Dunning, B.E.; Deacon, C.F.; Holst, J.J.; Foley, J.E. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 4888–4894. [Google Scholar] [CrossRef] [Green Version]
- Maiztegui, B.; Borelli, M.I.; Madrid, V.G.; Del Zotto, H.; Raschia, M.A.; Francini, F.; Massa, M.L.; Flores, L.E.; Rebolledo, O.R.; Gagliardino, J.J. Sitagliptin prevents the development of metabolic and hormonal disturbances, increased beta-cell apoptosis and liver steatosis induced by a fructose-rich diet in normal rats. Clin. Sci. 2011, 120, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haukeland, J.W.; Damas, J.K.; Konopski, Z.; Loberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjoro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Chen, Y.; Varghese, Z.; Ruan, X.Z. The molecular pathogenic role of inflammatory stress in dysregulation of lipid homeostasis and hepatic steatosis. Genes Dis. 2014, 1, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, M.M.; Ibrahim, R.M.; El Gazzar, W.B.; Said, M.A. Dipeptedyl peptidase-4 (DPP-4) inhibitor downregulates HMGB1/TLR4/NF-kappaB signaling pathway in a diabetic rat model of non-alcoholic fatty liver disease. Arch. Physiol. Biochem. 2021, 1–9. [Google Scholar] [CrossRef]
- Younan, N.; Elattar, S.; Farouk, M.; Rashed, L.; Estaphan, S. Dipeptidyl peptidase-4 inhibitors and aerobic exercise synergistically protect against liver injury in ovariectomized rats. Physiol. Rep. 2019, 7, e14191. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef]
- Angulo, P. Long-term mortality in nonalcoholic fatty liver disease: Is liver histology of any prognostic significance? Hepatology 2010, 51, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; McCullough, A.J. Noninvasive Diagnosis of NASH and Liver Fibrosis Within the Spectrum of NAFLD. Gastroenterol. Hepatol. 2012, 8, 661–668. [Google Scholar]
- Lakatos, P.L.; Firneisz, G.; Borcsiczky, D.; Zalatnai, A.; Selmeci, L.; Szalay, F. Elevated serum dipeptidyl peptidase IV (CD26, EC 3.4.14.5) activity in experimental liver cirrhosis. Eur. J. Clin. Investig. 2000, 30, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.L.; Firneisz, G.; Rakoczy, G.; Selmeci, L.; Szalay, F. Elevated serum dipeptidyl peptidase IV (CD26, EC 3.4.14.5) activity in patients with primary biliary cirrhosis. J. Hepatol. 1999, 30, 740. [Google Scholar] [CrossRef]
- Wang, X.M.; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; Vieira de Ribeiro, A.J.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Callanan, H.M.; Mowery, J.; Hixson, D.C. Evidence for a role of dipeptidyl peptidase IV in fibronectin-mediated interactions of hepatocytes with extracellular matrix. Biochem. J. 1989, 262, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.C.; Abdel-Ghany, M.; Pauli, B.U. A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J. Biol. Chem. 2003, 278, 24600–24607. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.O.; Schuppan, D. When GLP-1 hits the liver: A novel approach for insulin resistance and NASH. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G759–G761. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Thielitz, A.; Vetter, R.W.; Schultze, B.; Wrenger, S.; Simeoni, L.; Ansorge, S.; Neubert, K.; Faust, J.; Lindenlaub, P.; Gollnick, H.P.; et al. Inhibitors of dipeptidyl peptidase IV-like activity mediate antifibrotic effects in normal and keloid-derived skin fibroblasts. J. Investig. Dermatol. 2008, 128, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, N.N.; Li, Y.; Schuyler, C.A.; Lopes-Virella, M.F.; Huang, Y. DPP-4 (CD26) inhibitor alogliptin inhibits TLR4-mediated ERK activation and ERK-dependent MMP-1 expression by U937 histiocytes. Atherosclerosis 2010, 213, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salgado, C.; Fuentes-Calvo, I.; Garcia-Cenador, B.; Santos, E.; Lopez-Novoa, J.M. Involvement of H- and N-Ras isoforms in transforming growth factor-beta1-induced proliferation and in collagen and fibronectin synthesis. Exp. Cell Res. 2006, 312, 2093–2106. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willy, J.A.; Young, S.K.; Mosley, A.L.; Gawrieh, S.; Stevens, J.L.; Masuoka, H.C.; Wek, R.C. Function of inhibitor of Bruton’s tyrosine kinase isoform alpha (IBTKalpha) in nonalcoholic steatohepatitis links autophagy and the unfolded protein response. J. Biol. Chem. 2017, 292, 14050–14065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Lo, L.; McLennan, S.V.; Williams, P.F.; Bonner, J.; Chowdhury, S.; McCaughan, G.W.; Gorrell, M.D.; Yue, D.K.; Twigg, S.M. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J. Hepatol. 2011, 55, 435–444. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Ma, Q.; Lindsay, J.W.; Leone, K.A.; Ma, K.; Coben, J.; Galkina, E.V.; Nadler, J.L. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E410–E421. [Google Scholar] [CrossRef] [Green Version]
- Aston-Mourney, K.; Subramanian, S.L.; Zraika, S.; Samarasekera, T.; Meier, D.T.; Goldstein, L.C.; Hull, R.L. One year of sitagliptin treatment protects against islet amyloid-associated beta-cell loss and does not induce pancreatitis or pancreatic neoplasia in mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E475–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, W.L.; Wu, J.L.; Ren, B.R.; Zhang, H.Q. Hypoglycemic effects of a sesquiterpene glycoside isolated from leaves of loquat (Eriobotrya japonica (Thunb.) Lindl.). Phytomedicine Int. J. Phytother. Phytopharm. 2008, 15, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.J.; Yang, M.; Ren, J.W. Anti-diabetic and hypolipidemic effects of aqueous-extract from the flower of Inula japonica in alloxan-induced diabetic mice. Biol. Pharm. Bull. 2006, 29, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozera, B.; Rapacz, M. Reference genes in real-time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| MCP-1 | AGGTCCCTGTCATGCTTCTG | GCTGCTGGTGATCCTCTTGT |
| TNF-ɑ | CCCCAAAGGGATGAGAAGTT | CACTTGGTGGTTTGCTACGA |
| IL-1β | GACCTTCCAGGATGAGGACA | AGCTCATATGGGTCCGACAG |
| IL-6 | TTCACAAGTCCGGAGAGGAG | TTCTGCAAGTGCATCATCGT |
| Collagen I | CCCCGGGACTCCTGGACTT | GCTCCGACACGCCCTCTCTC |
| Collagen III | CCTGGAGCCCCTGGACTAATAG | GCCCATTGCACCAGGTTCT |
| Collagen IV-ɑ1 | ATCCGGCCCTTCATTAGC | ACTGCGGAATCTGAATGGTC |
| Collagen VI | GAACTTCCCTGCCAAACAGA | CACCTTGTGGAAGTTCTGCTC |
| CCN2 | GAAGGGCAAAAAGTGCATCC | CAGTTGTAATGGCAGGCAC |
| TGF-β1 | TGGAGCAACATGTGGAACTC | GTCAGCAGCCGGTTACCA |
| 18S | CGGCTACCACATACCAAGGAA | GCTGGAATTAACCGCGGCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Wang, X.; Yee, C.; Gorrell, M.D.; McLennan, S.V.; Twigg, S.M. Sitagliptin Is More Effective Than Gliclazide in Preventing Pro-Fibrotic and Pro-Inflammatory Changes in a Rodent Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Molecules 2022, 27, 727. https://doi.org/10.3390/molecules27030727
Ren J, Wang X, Yee C, Gorrell MD, McLennan SV, Twigg SM. Sitagliptin Is More Effective Than Gliclazide in Preventing Pro-Fibrotic and Pro-Inflammatory Changes in a Rodent Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Molecules. 2022; 27(3):727. https://doi.org/10.3390/molecules27030727
Chicago/Turabian StyleRen, Jing, Xiaoyu Wang, Christine Yee, Mark D. Gorrell, Susan V. McLennan, and Stephen M. Twigg. 2022. "Sitagliptin Is More Effective Than Gliclazide in Preventing Pro-Fibrotic and Pro-Inflammatory Changes in a Rodent Model of Diet-Induced Non-Alcoholic Fatty Liver Disease" Molecules 27, no. 3: 727. https://doi.org/10.3390/molecules27030727
APA StyleRen, J., Wang, X., Yee, C., Gorrell, M. D., McLennan, S. V., & Twigg, S. M. (2022). Sitagliptin Is More Effective Than Gliclazide in Preventing Pro-Fibrotic and Pro-Inflammatory Changes in a Rodent Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Molecules, 27(3), 727. https://doi.org/10.3390/molecules27030727