
Nucleophilic Substitution Reactions in the [B3H8]− Anion in the Presence of Lewis Acids

 ,
,  , ,
, ,  and
and
Abstract
:
1. Introduction
2. Experimental Procedure
2.1. Generation of [B3H7THF]
2.2. Generation of [B3H7NCCH3]
2.3. Generation of [B3H7NCCHPh2]
2.4. Synthesis of [B3H7PPh3]
2.5. Synthesis of [B3H7NEt3]
2.6. Generation of [B3H7 dppe] and [B3H7AsPh3]
2.7. Synthesis of [B3H7Py]
2.8. Synthesis of [B3H7NH2Ph]
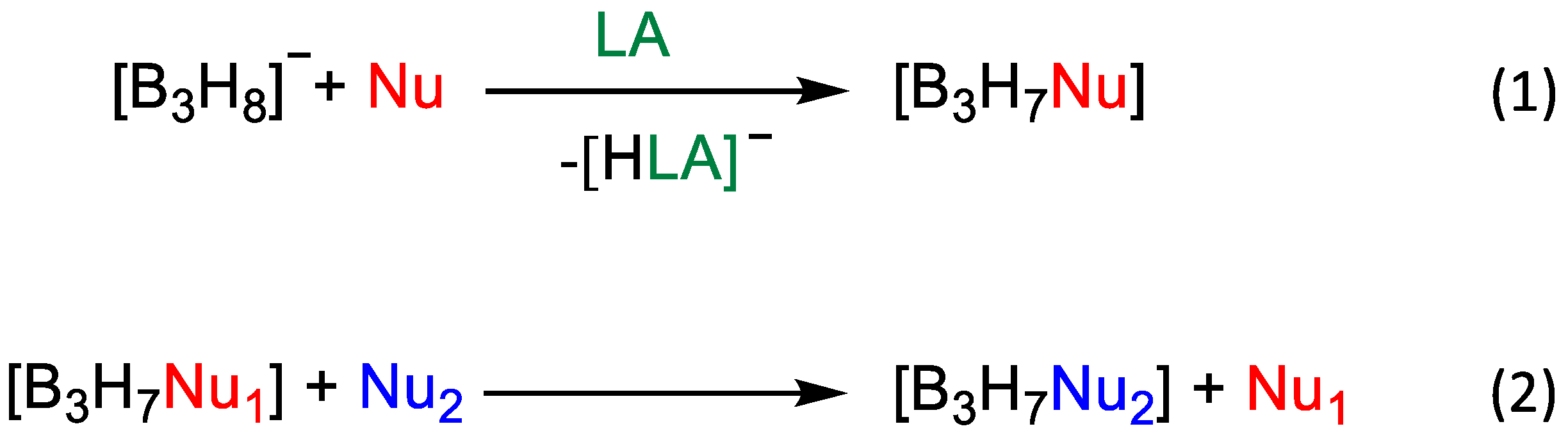
3. Results and Discussion
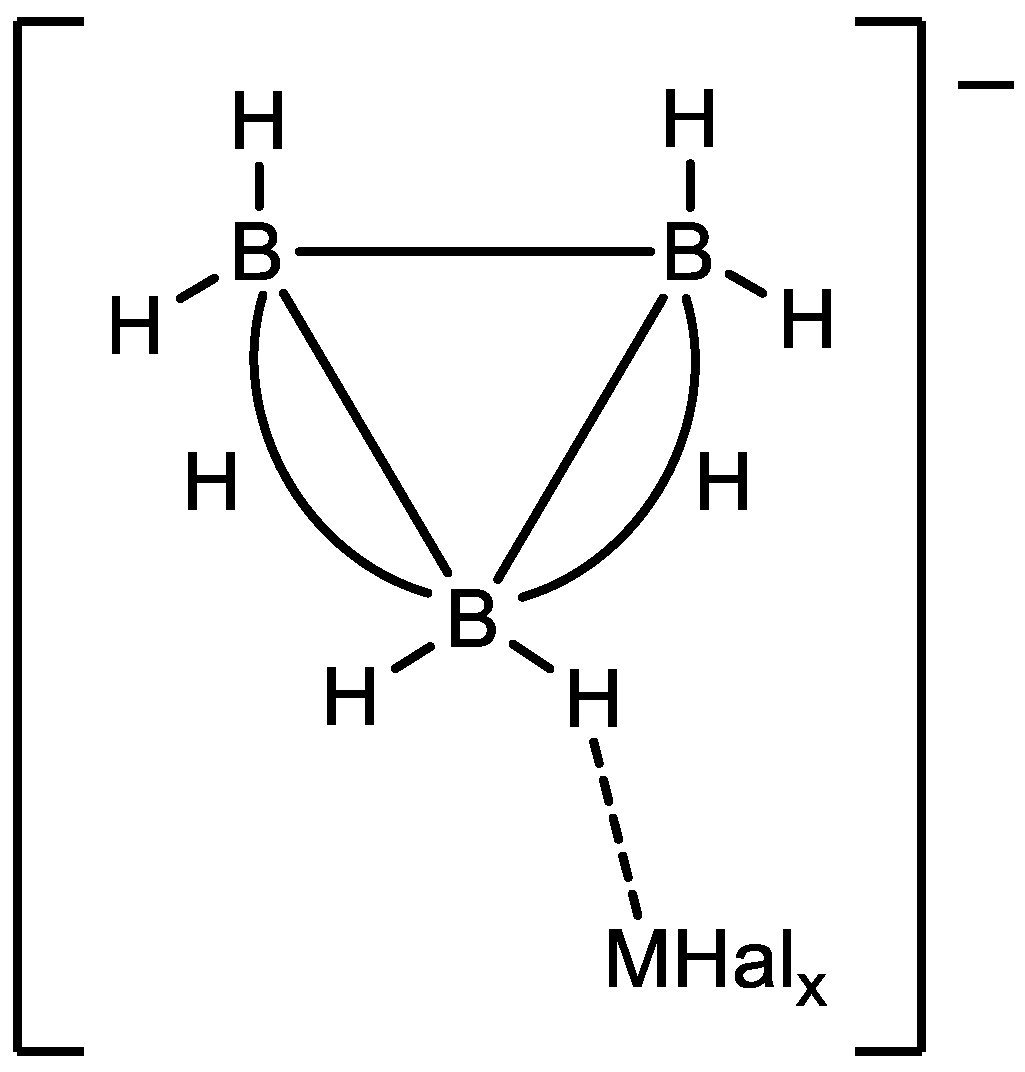
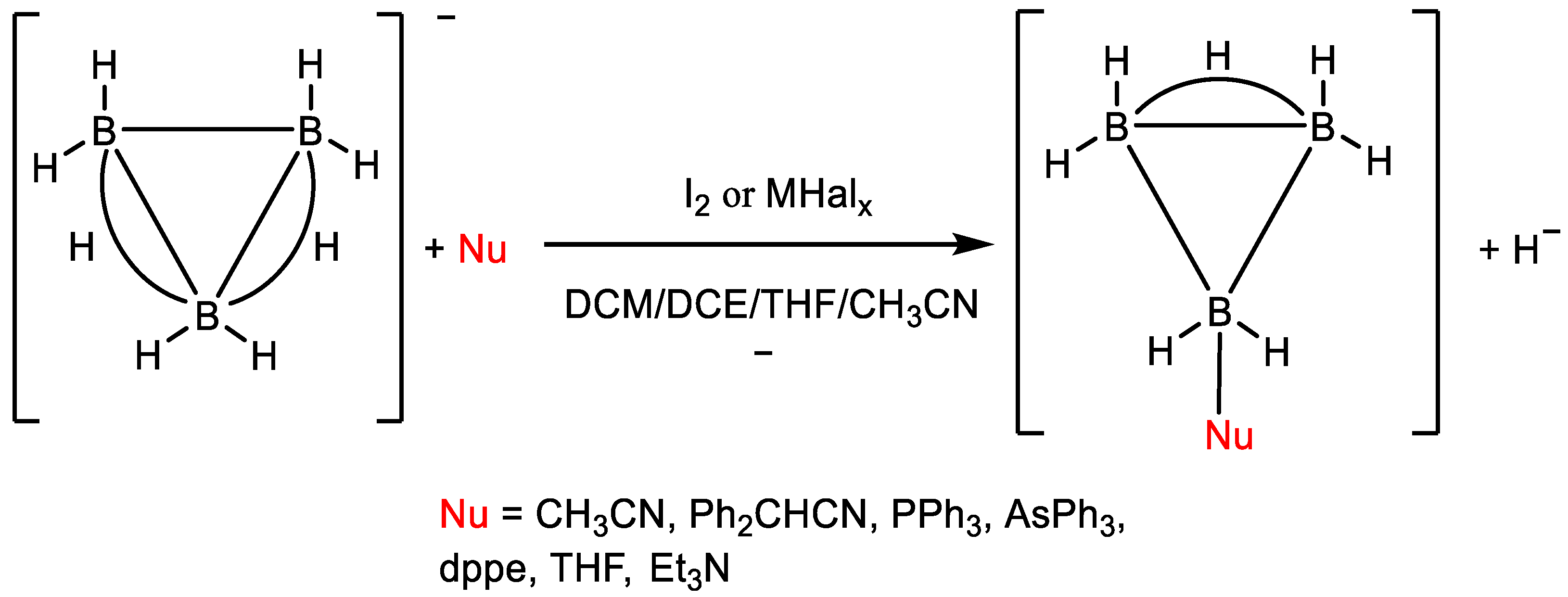
3.1. Interaction with Lewis Acids
3.2. Metathesis of Nucleophiles
3.3. Influence of Lewis Acids on the Evolution of the Reaction
3.4. NMR Data
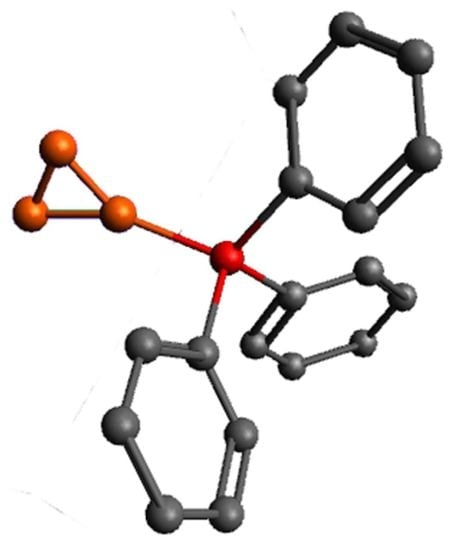
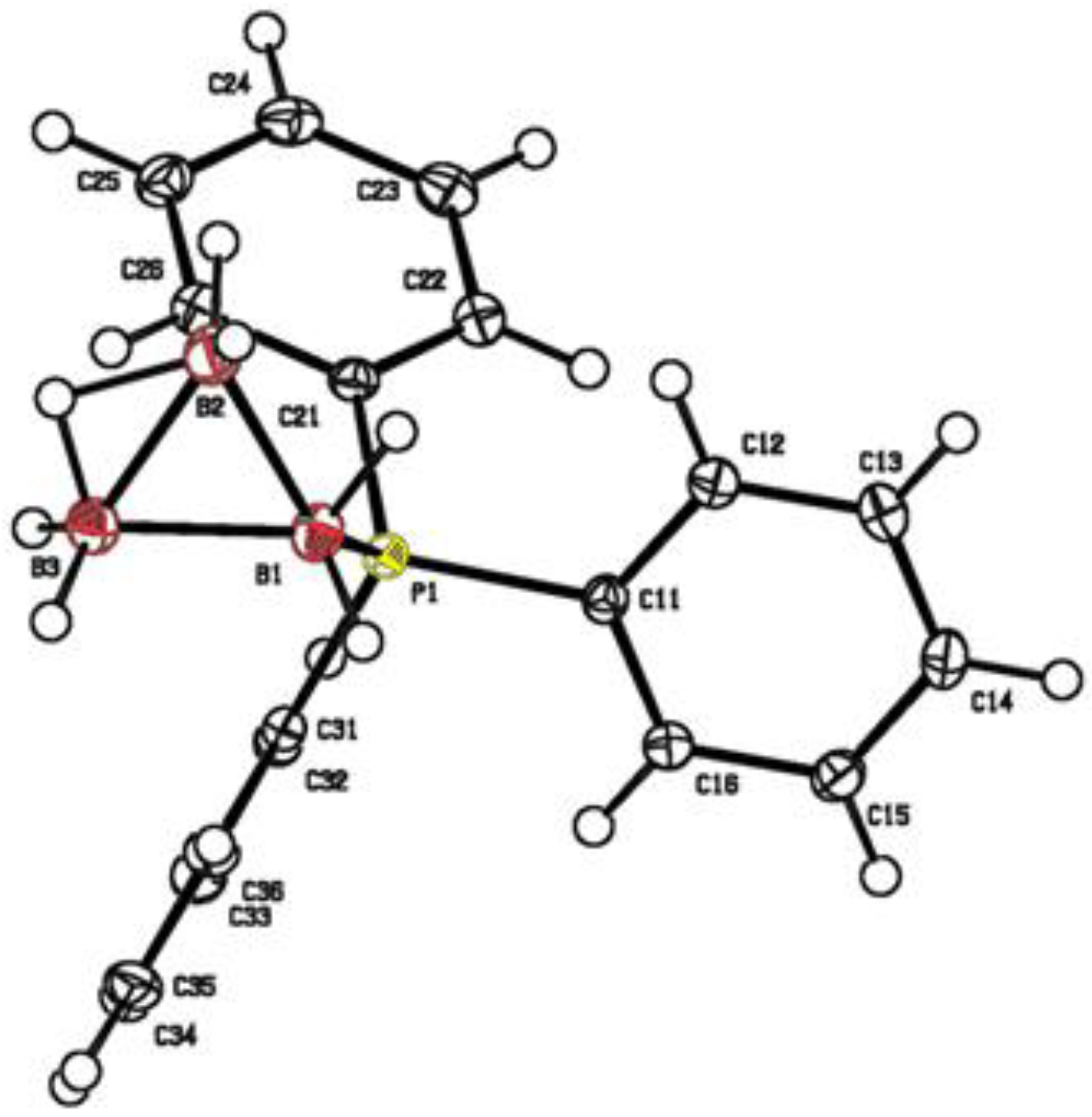
3.5. X-ray Diffraction Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Matveev, E.Y.; Kubasov, A.S.; Razgonyaeva, G.A.; Polyakova, I.N.; Zhizhin, K.Y.; Kuznetsov, N.T. Reactions of the [B10H10]2− anion with nucleophiles in the presence of halides of group IIIA and IVB elements. Russ. J. Inorg. Chem. 2015, 60, 776–785. [Google Scholar] [CrossRef]
- Zhizhin, K.Y.; Zhdanov, A.P.; Kuznetsov, N.T. Derivatives of closo-decaborate anion [B10H10]2− with exo-polyhedral substituents. Russ. J. Inorg. Chem. 2010, 55, 2089–2127. [Google Scholar] [CrossRef]
- Retivov, V.M.; Matveev, E.Y.; Lisovskiy, M.V.; Razgonyaeva, G.A.; Ochertyanova, L.I.; Zhizhin, K.Y.; Kuznetsov, N.T. Nucleophilic substitution in closo-decaborate [B10H10]2− in the presence of carbocations. Russ. Chem. Bull. 2010, 59, 1–6. [Google Scholar] [CrossRef]
- Mu, X.; Axtell, J.C.; Bernier, N.A.; Kirlikovali, K.O.; Jung, D.; Umanzor, A.; Qian, K.; Chen, X.; Bay, K.L.; Kirollos, M.; et al. Sterically Unprotected Nucleophilic Boron Cluster Reagents. Chem 2019, 5, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, G.B.; Morris, J.H. Preparation of substituted derivatives of the octahydrotriborate(1−) ion. Inorg. Chim. Acta 1982, 59, 207–211. [Google Scholar] [CrossRef]
- Davis, C.M.; Hamilton, G.A.; Schnee, V.P. Hydride abstraction from the octahydrotriborate ion and formation of bridged triborane(7)-phosphane complexes. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 1641–1644. [Google Scholar] [CrossRef]
- Yoon, C.W.; Carroll, P.J.; Sneddon, L.G. Ammonia triborane: A new synthesis, structural determinations, and hydrolytic hydrogen-release properties. J. Am. Chem. Soc. 2009, 131, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M.; Ma, N.; Zhang, Q.-F.; Wang, J.; Feng, X.; Wei, C.; Wang, L.-S.; Zhang, J.; Chen, X. Elucidation of the Formation Mechanisms of the Octahydrotriborate Anion (B3H8−) through the Nucleophilicity of the B-H Bond. J. Am. Chem. Soc. 2018, 140, 6718–6726. [Google Scholar] [CrossRef] [PubMed]
- Goedde, D.M.; Girolami, G.S. A new class of CVD precursors to metal borides: Cr(B3H8)2 and related octahydrotriborate complexes. J. Am. Chem. Soc. 2004, 126, 12230–12231. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Møller, K.T.; Yan, Y.; Chen, X.-M.; Li, Y.; Li, H.-W.; Zhou, W.; Skibsted, J.; Chen, X.; Jensen, T.R. Potassium octahydridotriborate: Diverse polymorphism in a potential hydrogen storage material and potassium ion conductor. Dalt. Trans. 2019, 48, 8872–8881. [Google Scholar] [CrossRef] [PubMed]
- Moussa, G.; Moury, R.; Demirci, U.B.; Sener, T.M.P. Boron-based hydrides for chemical hydrogen storage. Int. J. Energy Res. 2013, 37, 825–842. [Google Scholar] [CrossRef]
- Huang, Z.; Eagles, M.; Porter, S.; Sorte, E.G.; Billet, B.; Corey, R.L.; Conradi, M.S.; Zhao, J.-C. Thermolysis and solid state NMR studies of NaB3H8, NH3B3H7, and NH4B3H8. Dalt. Trans. 2013, 42, 701–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.R.; Paskevicius, M.; Hansen, B.R.; Jakobsen, A.S.; Møller, K.T.; White, J.L.; Allendorf, M.D.; Stavila, V.; Skibsted, J.; Jensen, T.R. Hydrogenation properties of lithium and sodium hydride- closo -borate, [B10H10]2− and [B12H12]2−, composites. Phys. Chem. Chem. Phys. 2018, 20, 16266–16275. [Google Scholar] [CrossRef] [Green Version]
- Gaines, D.F.; Hildebrandt, S.J. Syntheses and Properties of Some Neutral Octahydrotriborate(1-) Complexes of Chromium-, Manganese-, and Iron-Group Metals. Inorg. Chem. 1978, 17, 794–806. [Google Scholar] [CrossRef]
- Malinina, E.A.; Korolenko, S.E.; Goeva, L.V.; Buzanov, G.A.; Avdeeva, V.V.; Kuznetsov, N.T. Synthesis and Structure of [M(DMF)6][B10H10] (M = Zn(II), Cd(II)) as Precursors for Solid-Phase Synthesis of Trischelate Complexes [M(L)3][B10H10]. Russ. J. Inorg. Chem. 2018, 63, 1552–1557. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, Y.; Li, M.; Zhao, F.; Gu, Q.; Ma, X.; Guo, Y. Li(NH3)B3H8: A new ionic liquid octahydrotriborate. Chem. Commun. 2019, 55, 408–411. [Google Scholar] [CrossRef]
- Kher, S.S.; Romero, J.V.; Caruso, J.D.; Spencer, J.T. Chemical vapor deposition of metal borides: 6. the formation of neodymium boride thin film materials from polyhedral boron clusters and metal halides by chemical vapor deposition. Appl. Organomet. Chem. 2008, 22, 300–307. [Google Scholar] [CrossRef]
- Chen, H.; Zou, X. Intermetallic borides: Structures, synthesis and applications in electrocatalysis. Inorg. Chem. Front. 2020, 7, 2248–2264. [Google Scholar] [CrossRef]
- Jayaraman, S.; Klein, E.J.; Yang, Y.; Kim, D.Y.; Girolami, G.S.; Abelson, J.R. Chromium diboride thin films by low temperature chemical vapor deposition. J. Vac. Sci. Technol. A Vac. Surf. Film. 2005, 23, 631–633. [Google Scholar] [CrossRef]
- Ivanov, B.L.; Wellons, M.S.; Lukehart, C.M. Confined-plume chemical deposition: Rapid synthesis of crystalline coatings of known hard or superhard materials on inorganic or organic supports by resonant IR decomposition of molecular precursors. J. Am. Chem. Soc. 2009, 131, 11744–11750. [Google Scholar] [CrossRef] [PubMed]
- Auerhammer, D.; Arrowsmith, M.; Braunschweig, H.; Dewhurst, R.D.; Jiménez-Halla JO, C.; Kupfer, T. Nucleophilic addition and substitution at coordinatively saturated boron by facile 1,2-hydrogen shuttling onto a carbene donor. Chem. Sci. 2017, 8, 7066–7071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.; Morris, J.H. Reactions of the octahydrotriborate (−1) ion with mercury salts. Inorg. Chim. Acta 1977, 24, 191–194. [Google Scholar] [CrossRef]
- Sethio, D.; Lawson Daku, L.M.; Hagemann, H. A theoretical study of the spectroscopic properties of B2H6 and of a series of Bx Hyz- species (x = 1–12, y = 3–14, z = 0–2): From BH3 to B12H122−. Int. J. Hydrogen Energy 2016, 41, 6814–6824. [Google Scholar] [CrossRef] [Green Version]
- Bailey, W.J.; Marktscheffel, F. Cleavage of Tetrahydrofuran during Reductions with Lithium Aluminum Hydride. J. Org. Chem. 1960, 25, 1797–1800. [Google Scholar] [CrossRef]
- Serrar, C.; Ouassas, A. Synthesis, Reactivity and Theoretical Study of B3H8− and Related Derivatives. Synth. React. Inorg. Met. Org. Chem. 1996, 26, 669–683. [Google Scholar] [CrossRef]
- Parry, R.W.; Edwards, L.J. Systematics in the Chemistry of the Boron Hydrides. J. Am. Chem. Soc. 1959, 81, 3554–3560. [Google Scholar] [CrossRef]
- Ishii, M.; Kodama, G. Reaction of Dimethyl Sulfide-Triborane with Trimethylamine. Facile Formation of Bis(trimethylamine)—Diborane. Inorg. Chem. 1990, 29, 2181–2183. [Google Scholar] [CrossRef]
- Ishii, M.; Kodama, G. Reaction of Dimethyl Sulfide-Triborane(7) with Dimethyl Sulfide. A Formation Reaction of Pentaborane(9). Inorg. Chem. 1990, 29, 817–820. [Google Scholar] [CrossRef]
- Graybill, M.; Ruff, J.K. Cleavage of Trimethylamine Triborane. J. Am. Chem. Soc. 1962, 84, 1062–1063. [Google Scholar] [CrossRef]
- Matveev, E.Y.; Akimov, S.S.; Kubasov, A.S.; Retivov, V.M.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis and Study of Derivatives of the [B10H10]2– Anion with Amino Acids. Russ. J. Inorg. Chem. 2019, 64, 1513–1521. [Google Scholar] [CrossRef]
- DePoy, R.; Kodama, G. Isolation and characterization of bis(trimethylamine)-diborane(4). Inorg. Chem. 1985, 24, 2871–2872. [Google Scholar] [CrossRef]
- Kodama, G.; Parry, R.W.; Carter, J.C. The Preparation and Properties of Ammonia-Triborane, H3NB3H7. J. Am. Chem. Soc. 1959, 81, 3534–3538. [Google Scholar] [CrossRef]
- Polyakova, I.N.; Malinina, E.A.; Kuznetsov, N.T. Crystal structures of cesium and dimethylammonium cupradecaborates, Cs[CuB10H10] and (CH3)2NH2[CuB10H10]. Kristallografiya 2003, 48, 89–96. [Google Scholar]
- Schaeffer, R. Interconversion of boranes—III. An analysis of the “first stable intermediate” problem. J. Inorg. Nucl. Chem. 1960, 15, 190–193. [Google Scholar] [CrossRef]
- Tebbe, F.N. Synthesis of a Boron-Labeled Tetraborane. J. Am. Chem. Soc. 1962, 84, 3974–3975. [Google Scholar]
- Gao, P.; Wang, X.; Huang, Z.; Yu, H. 11B NMR chemical shift predictions via density functional theory and gauge-including atomic orbital approach: Applications to structural elucidations of boron-containing molecules. ACS Omega 2019, 4, 12385–12392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.W.; Calabrese, J.C.; Gaines, D.F.; Hillenbrand, D.F. Low-temperature crystal and molecular structure of tetracarbonyl [2-bromoheptahydrotriborato (1−)] manganese,(CO) 4MnB3H7Br, and a proton NMR study of the kinetics of its intramolecular hydrogen exchange in solution. J. Am. Chem. Soc. 1980, 102, 4928–4933. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brassington, D.S.; Coles, S.J.; Gelbrich, T.; Hursthouse, M.B. Synthesis and characterisation of a series of Group 7 metal 2,2,2,2-dicarbonylbis(triorganophosphine)-arachno-2-metallatetraboranes, [M(CO)2L2(B3H8)] (M = Re, Mn); crystal and molecular structures of [Re(CO)2(dppf)(B3H8)] and [Mn(CO)2(dppe)(B3H8)]. Polyhedron 2003, 22, 1627–1632. [Google Scholar] [CrossRef]
- Kim, D.Y.; Girolami, G.S. Synthesis and characterization of the octahydrotriborate complexes Cp*V(B3H8)2 and Cp*Cr(B3H8)2 and the unusual cobaltaborane cluster Cp*2Co2(B6H14). J. Am. Chem. Soc. 2006, 128, 10969–10977. [Google Scholar] [CrossRef]
- Liu, X.R.; Chen, X.M.; Zhang, J.; Jensen, T.R.; Chen, X. The interconversion between THF·B3H7 and B3H8−: An efficient synthetic method for MB3H8 (M = Li and Na). Dalt. Trans. 2019, 48, 5140–5143. [Google Scholar] [CrossRef] [PubMed]
- Dodds, A.R.; Kodama, G. A Comparative Study of Methylamine Adducts of Triborane(7). Inorg. Chem. 1976, 15, 741–743. [Google Scholar] [CrossRef]
- Nordman, C.E.; Reimann, C. The Molecular and Crystal Structures of Ammonia-Triborane 1. J. Am. Chem. Soc. 1959, 81, 3538–3543. [Google Scholar] [CrossRef]
- Andrews, S.J.; Welch, A.J. The structure of methylenetriphenylphosphorane(C–B)triborane(7) at 185 K. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1985, 41, 1496–1499. [Google Scholar] [CrossRef]
- Andrews, S.J.; Welch, A.J. Molecular and crystal structures of [(Ph3P)2N] [B3H7(NCS)] and [(Ph3P)2N] [B3H7(NCSe)]; the need for low-temperature x-ray crystallography. Inorg. Chim. Acta 1984, 88, 153–160. [Google Scholar] [CrossRef]
- Ghanta, S.R.; Rao, M.H.; Muralidharan, K. Single-pot synthesis of zinc nanoparticles, borane (BH3) and closo-dodecaborate (B12H12)2− using LiBH4 under mild conditions. Dalt. Trans. 2013, 42, 8420. [Google Scholar] [CrossRef]
- Funnell, N.P.; Dawson, A.; Marshall, W.G.; Parsons, S. Destabilisation of hydrogen bonding and the phase stability of aniline at high pressure. CrystEngComm 2013, 15, 1047–1060. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Ph3P·B3H7 | PhNH2·B3H7 |
|---|---|---|
| B–P(N) | 1.926(1) | 1.608(3) |
| B1–B2(B3) | 1.804(2) 1.821(2) | 1.847(3) 1.830(3) |
| B2–B3 | 1.779(2) | 1.719(4) |
| B2–Hbr | 1.33(2) | 1.32(4) |
| B3–Hbr | 1.17(2) | 1.27(4) |
| av. B1–H | 1.10 | 1.12 |
| av. B2(3)–Hterm | 1.11 | 1.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulyak, A.T.; Bortnikov, E.O.; Selivanov, N.A.; Grigoriev, M.S.; Kubasov, A.S.; Zhdanov, A.P.; Bykov, A.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. Nucleophilic Substitution Reactions in the [B3H8]− Anion in the Presence of Lewis Acids. Molecules 2022, 27, 746. https://doi.org/10.3390/molecules27030746
Shulyak AT, Bortnikov EO, Selivanov NA, Grigoriev MS, Kubasov AS, Zhdanov AP, Bykov AY, Zhizhin KY, Kuznetsov NT. Nucleophilic Substitution Reactions in the [B3H8]− Anion in the Presence of Lewis Acids. Molecules. 2022; 27(3):746. https://doi.org/10.3390/molecules27030746
Chicago/Turabian StyleShulyak, Alexandra T., Evgeniy O. Bortnikov, Nikita A. Selivanov, Mikhail S. Grigoriev, Alexey S. Kubasov, Andrey P. Zhdanov, Alexander Y. Bykov, Konstantin Y. Zhizhin, and Nikolai T. Kuznetsov. 2022. "Nucleophilic Substitution Reactions in the [B3H8]− Anion in the Presence of Lewis Acids" Molecules 27, no. 3: 746. https://doi.org/10.3390/molecules27030746
APA StyleShulyak, A. T., Bortnikov, E. O., Selivanov, N. A., Grigoriev, M. S., Kubasov, A. S., Zhdanov, A. P., Bykov, A. Y., Zhizhin, K. Y., & Kuznetsov, N. T. (2022). Nucleophilic Substitution Reactions in the [B3H8]− Anion in the Presence of Lewis Acids. Molecules, 27(3), 746. https://doi.org/10.3390/molecules27030746