
Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents

 , and
, and
Abstract
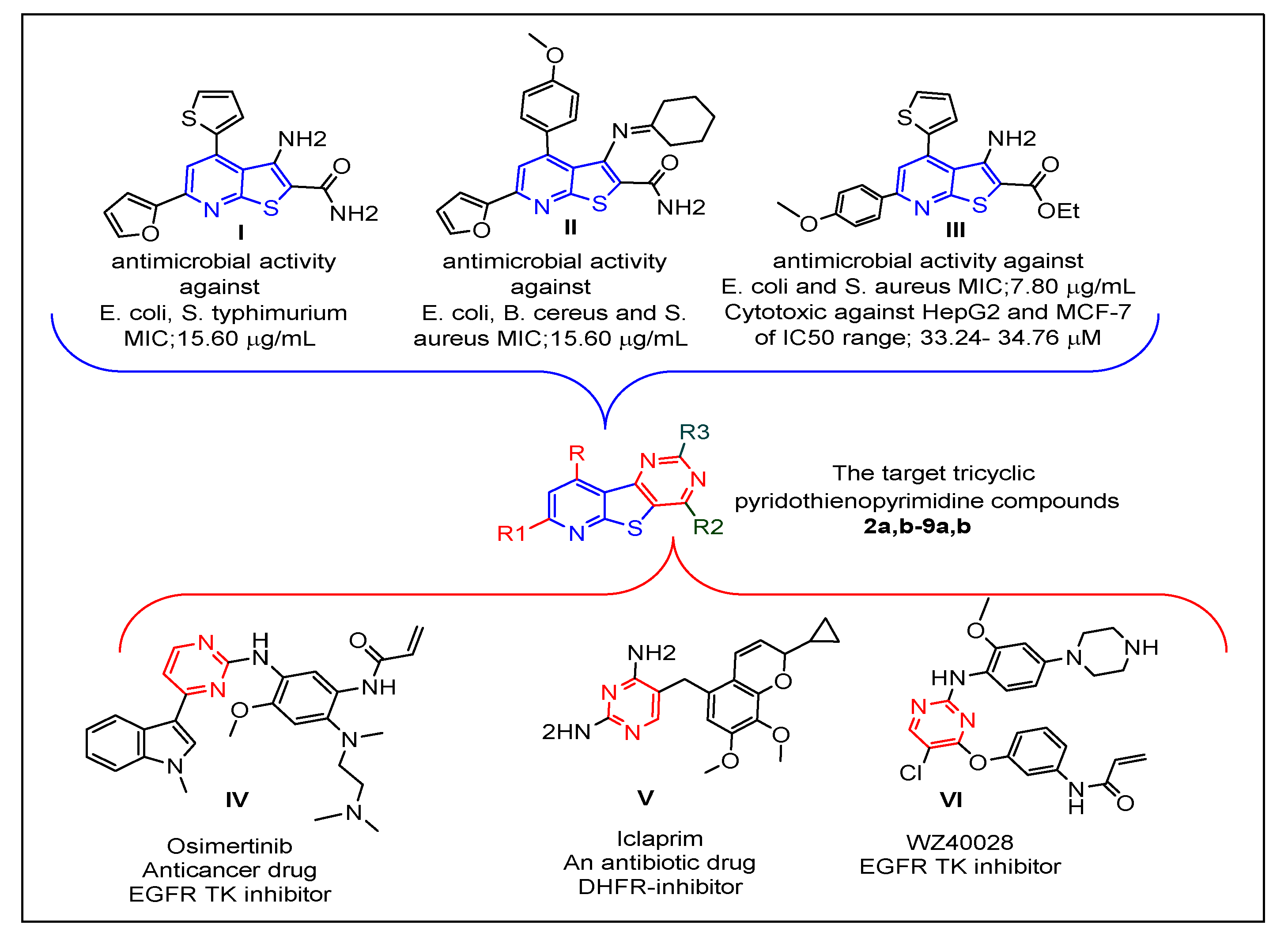
:1. Introduction
2. Results and Discussion
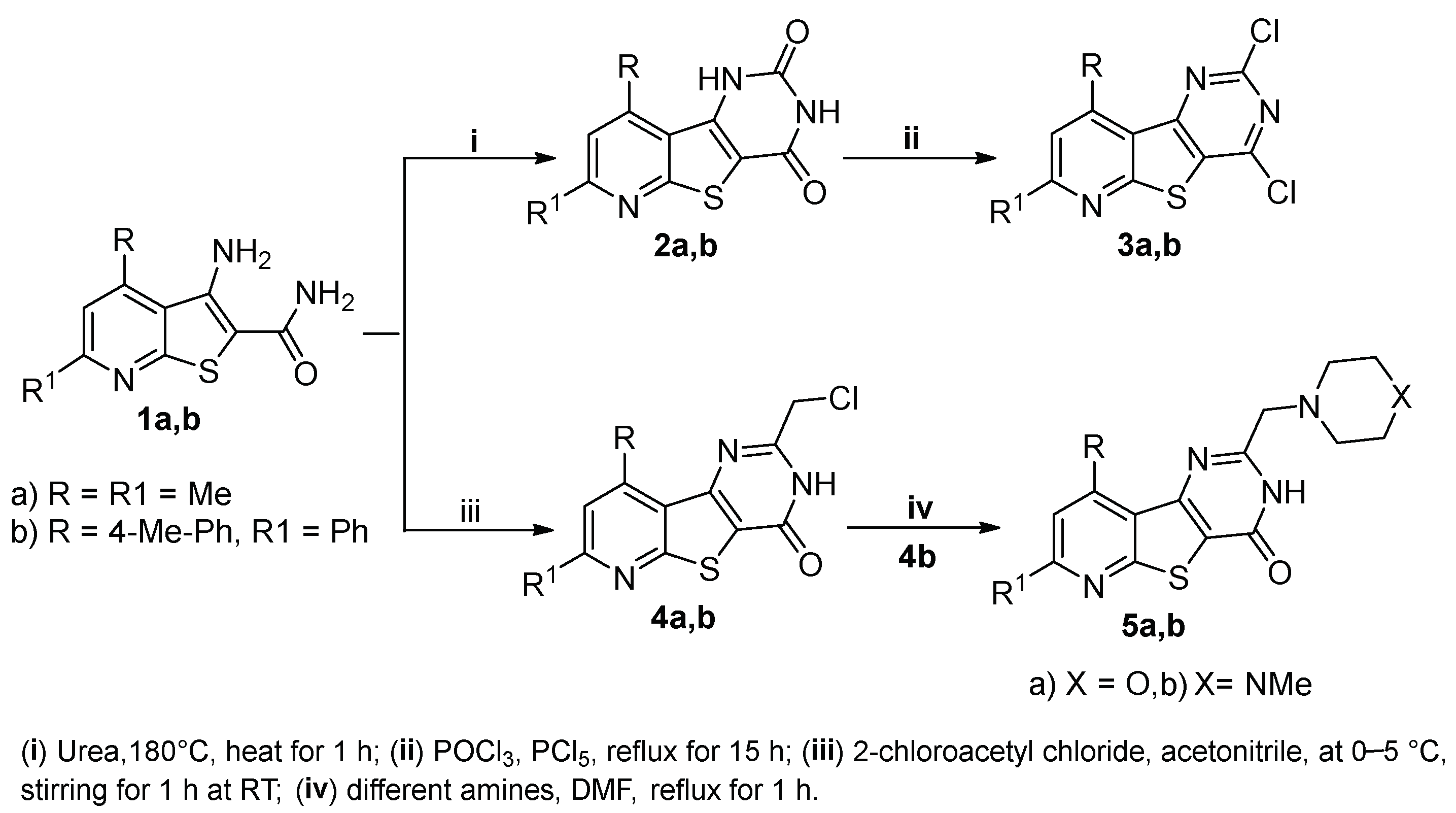
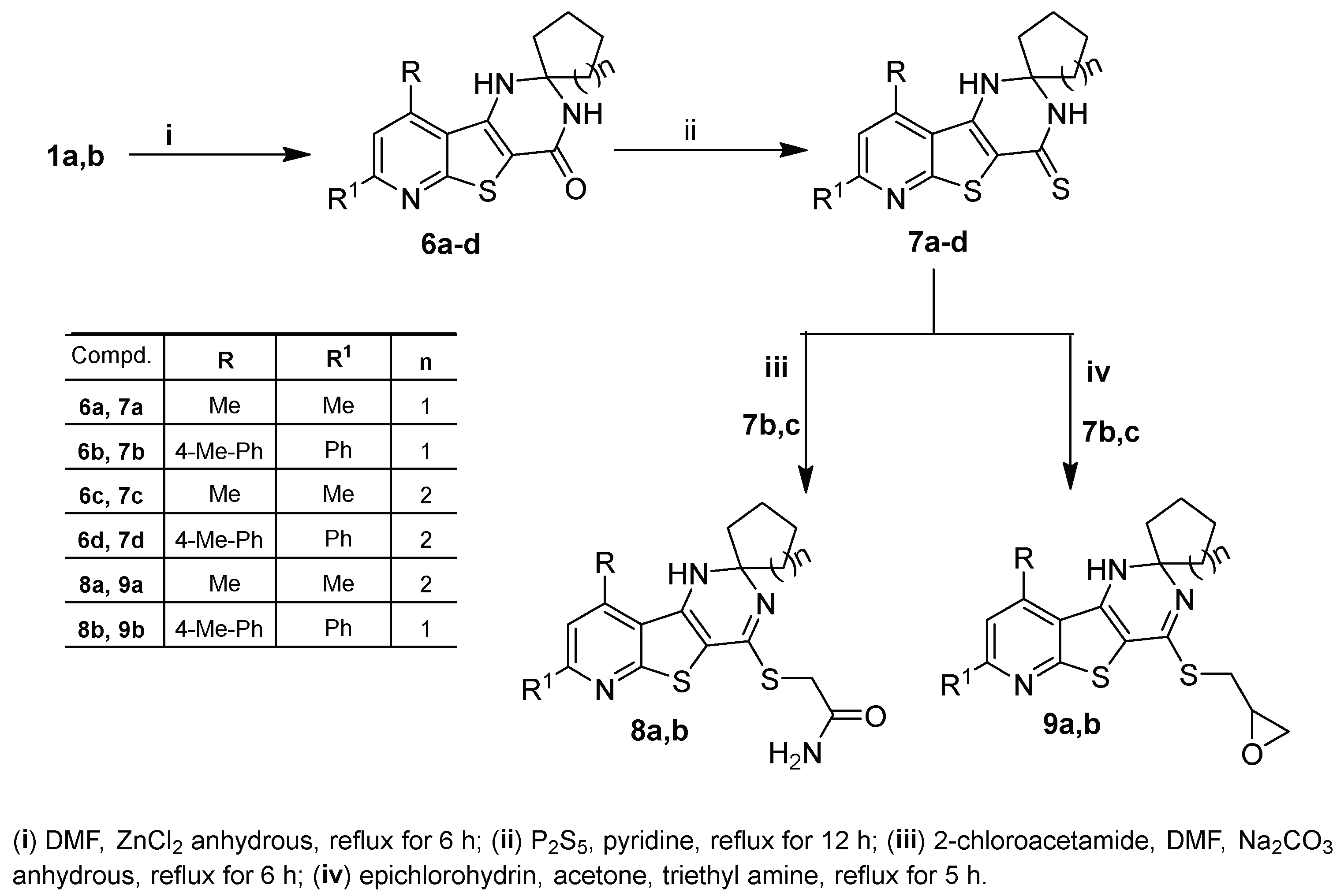
2.1. Chemistry
2.2. Biological Activity
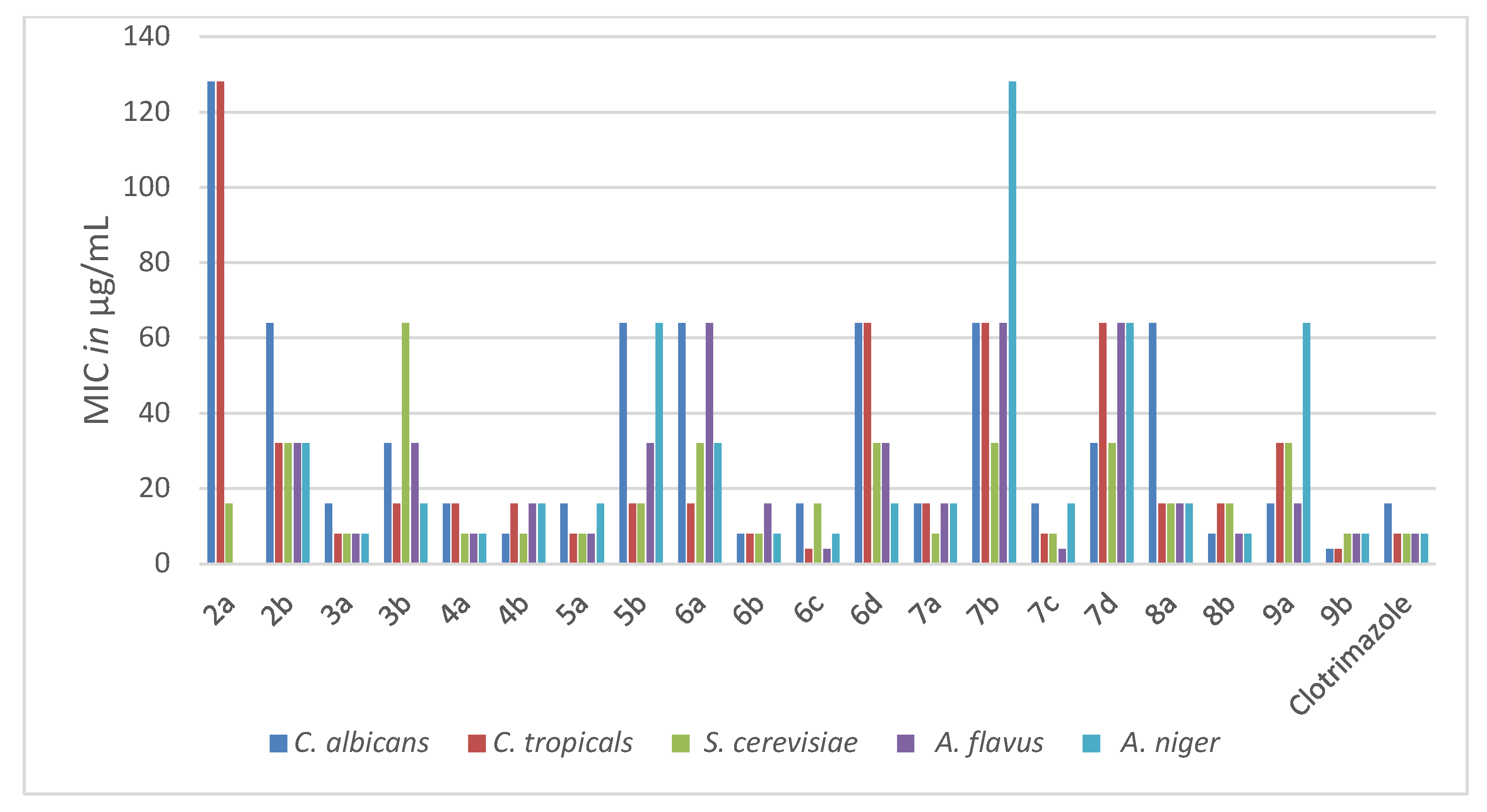
2.2.1. Antimicrobial Activity
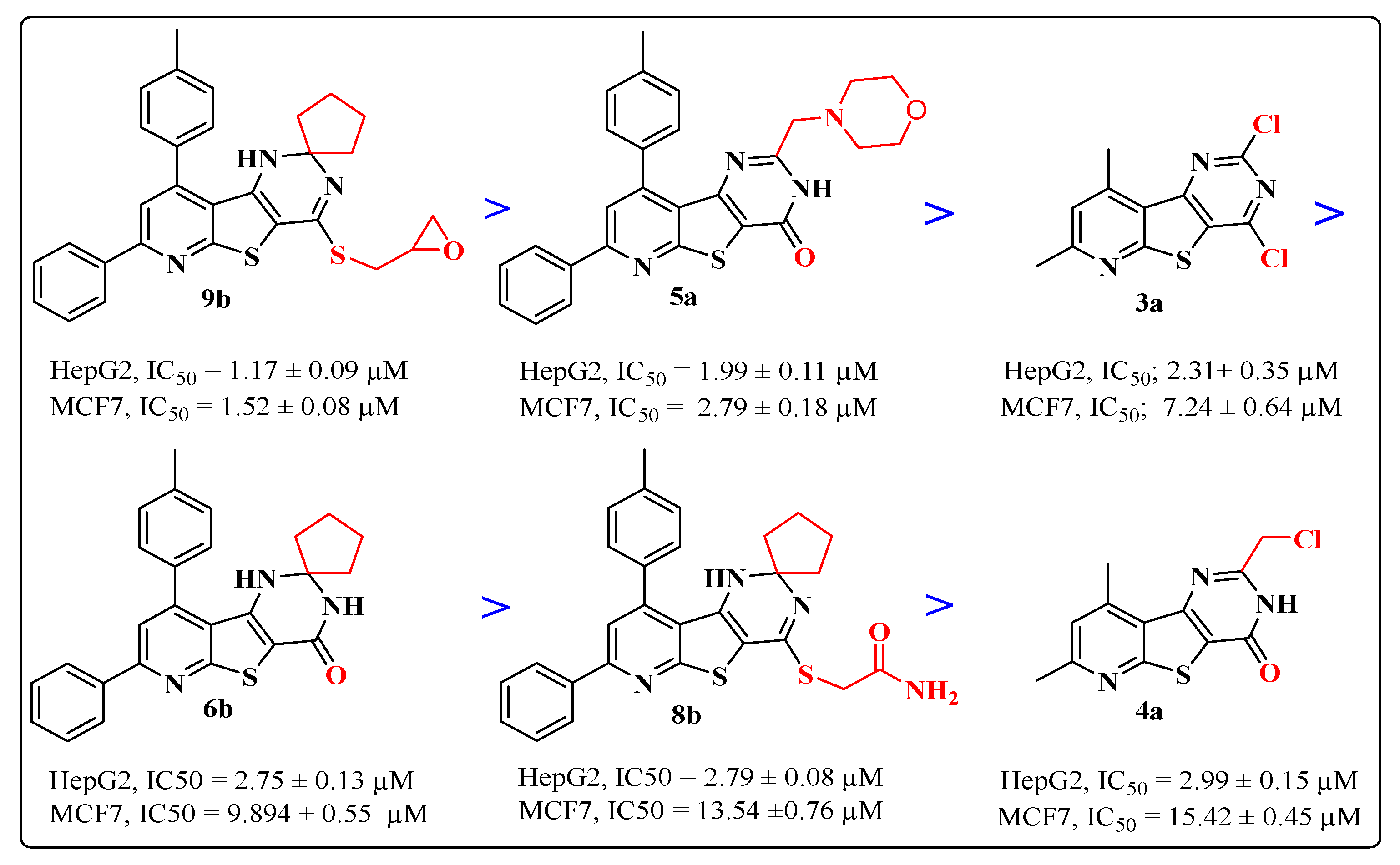
2.2.2. Cytotoxic Activity
2.2.3. In Vitro EGFR Enzyme Inhibition Assay
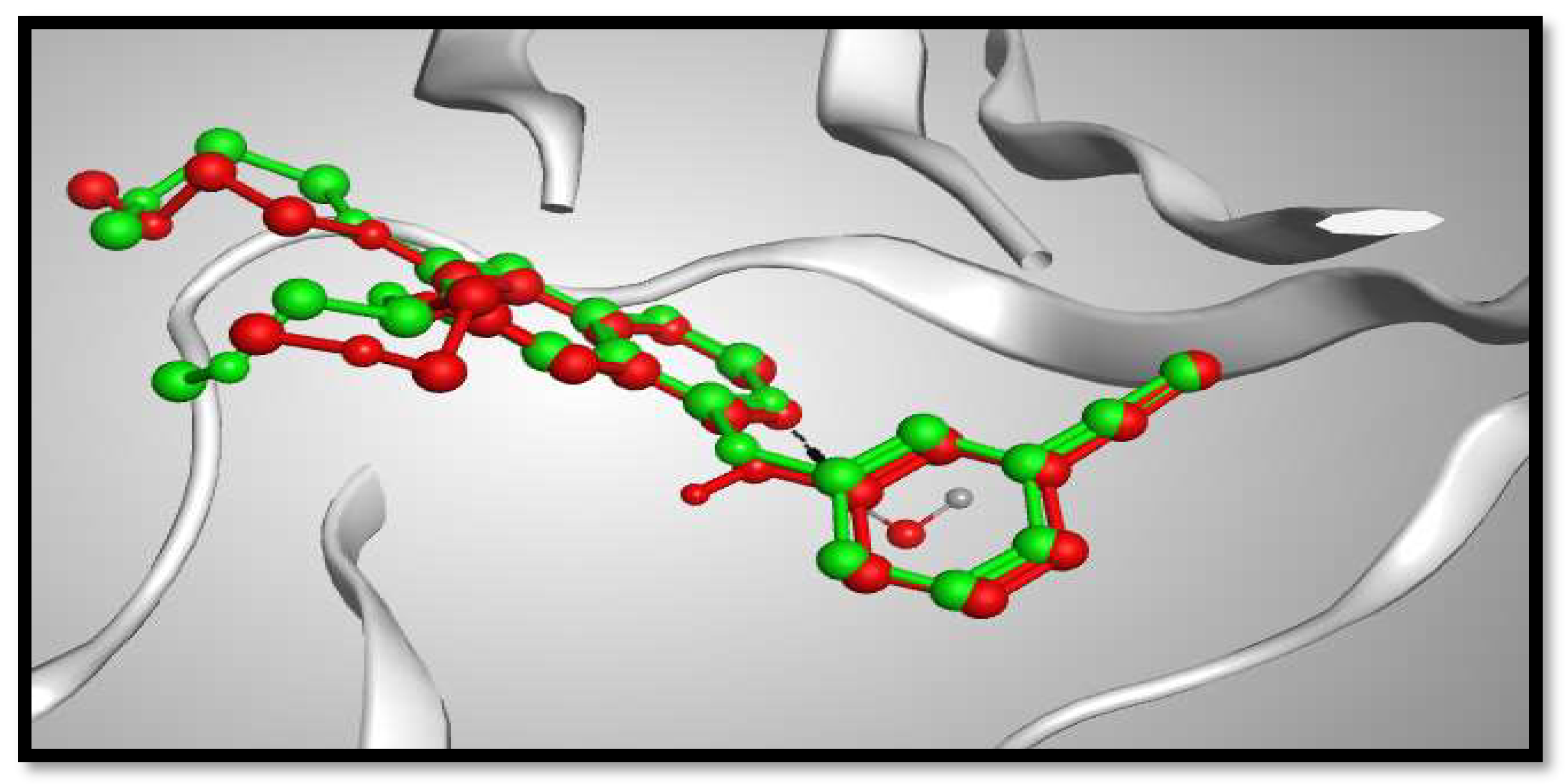
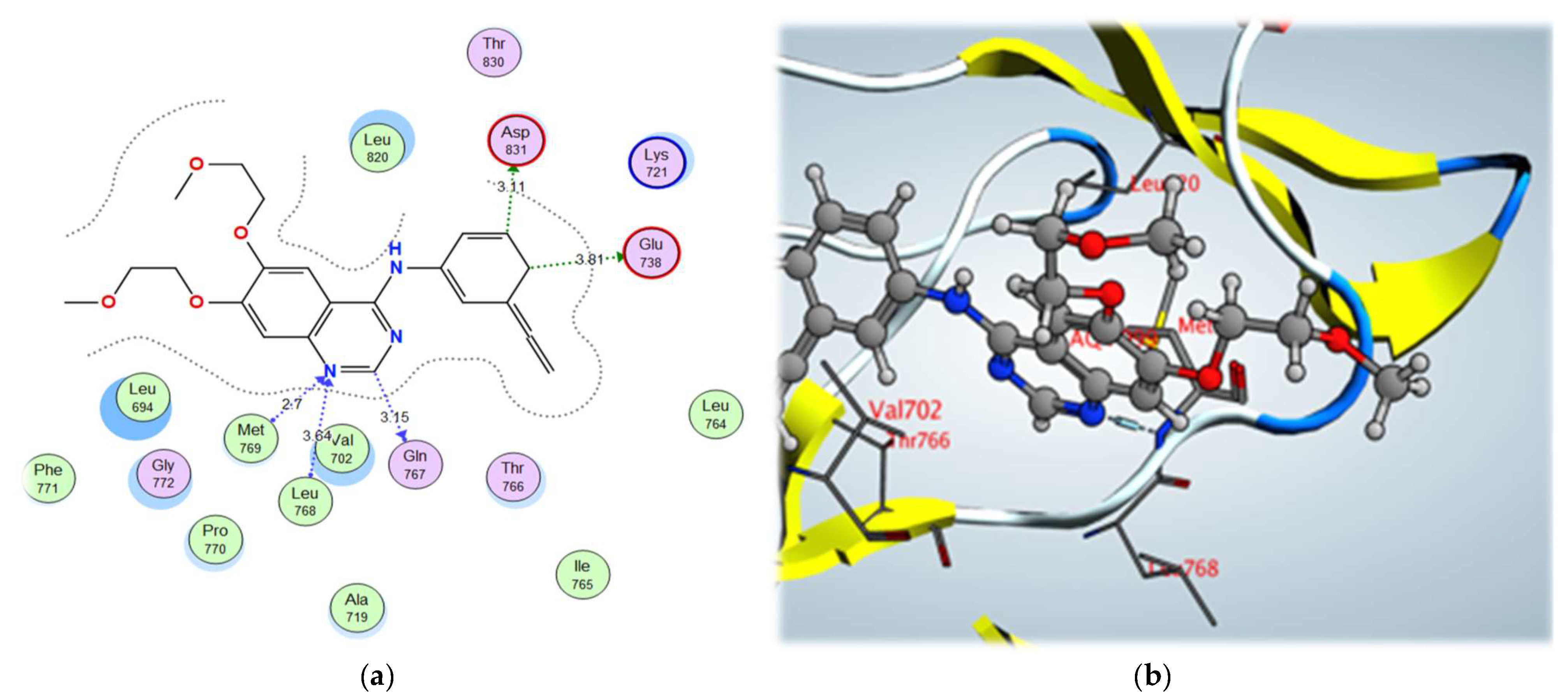
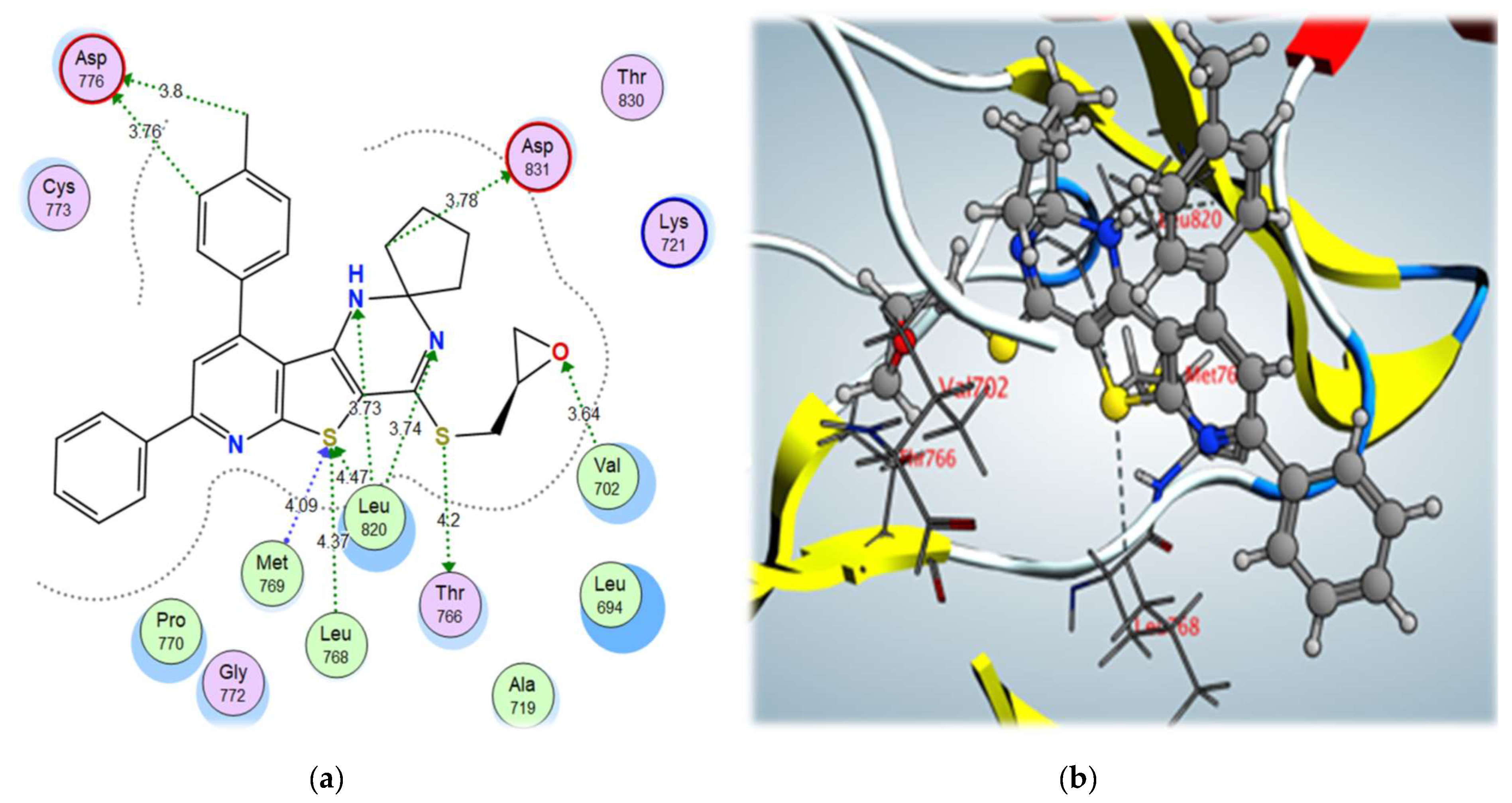
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of Pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-diones 2a,b
3.1.3. Synthesis of 2,4-Dichloropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines 3a,b
3.1.4. Synthesis of 2-(Chloromethyl)pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(3H)-ones 4a,b
3.1.5. Synthesis of 2-Substituted-7-phenyl-9-(p-tolyl)pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(3H)-ones 5a,b
3.1.6. Synthesis of 1′H-Spiro[cycloalkane-1,2′-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin]-4′(3′H)-ones 6a–d
3.1.7. Synthesis of 1′H-Spiro[cycloalkane-1,2′-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine]-4′(3′H)-thiones 7a–d
3.1.8. Synthesis of 2-((1′H-Spiro[cycloalkane-1,2′-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin]-4′-yl)sulfanyl)acetamides 8a,b
3.1.9. Synthesis of 4′-((oxiran-2-ylmethyl)sulfanyl)-1′H-spiro[cycloalkane-1,2′-pyrido[3′,2′:4,5]thieno-[3,2-d]pyrimidine] 9a,b
3.2. Antimicrobial Assay
3.3. In Vitro Cytotoxicity Screening
3.4. EGFR Kinase Inhibitory Assay
3.5. Molecular Modeling Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hay, S.I.; Rao, P.C.; Dolecek, C.; Day, N.P.J.; Stergachis, A.; Lopez, A.D.; Murray, C.J.L. Measuring and mapping the global burden of antimicrobial resistance. BMC Med. 2018, 16, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomazzi, M.; Moore, M.; Johnson, A.; Balasegaram, M.; Borisch, B. Antimicrobial resistance—Moving forward? BMC Public Health 2019, 19, 858. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.F.; Stephen, J.; Lekshmi, M.; Ojha, M.; Wenzel, N.; Sanford, L.M.; Hernandez, A.J.; Parvathi, A.; Kumar, S.H. Bacterial Resistance to Antimicrobial Agents. Antibiotics 2021, 10, 593. [Google Scholar] [CrossRef]
- Annunziato, G. Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review. Int. J. Mol. Sci. 2019, 20, 5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Jiang, D.; Hu, G. The antibacterial activity of isatin hybrids. Curr. Top Med. Chem. 2021, 22, 25–40. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kaur, S.; Chadha, B.S.; Kaur, R.; Kaur, M.; Kaur, S. Anticancer and antimicrobial potential of enterocin 12a from Enterococcus faecium. BMC Microbiol. 2021, 21, 39. [Google Scholar] [CrossRef]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with dual antimicrobial and anticancer activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinzadeh, Z.; Ramazani, A.; Razzaghi-Asl, N. Anti-cancer nitrogen-containing heterocyclic compounds. Curr. Org. Chem. 2018, 22, 2256–2279. [Google Scholar] [CrossRef]
- Mermer, A.; Keles, T.; Sirin, Y. Recent Studies of Nitrogen Containing Heterocyclic Compounds as Novel Antiviral Agents: A Review. Bioorganic Chem. 2021, 114, 105076. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, X.H.; Wang, S.Q.; Zhao, W.; Chen, X.B.; Yua, B. FDA-approved pyrimidine-fused bicyclic heterocycles for cancer therapy: Synthesis and clinical application. Eur. J. Med. Chem. 2021, 2141, 13218. [Google Scholar] [CrossRef]
- Mohi El-Deen, E.M.; Anwar, M.M.; Hasabelnaby, S.M. Synthesis and in vitro cytotoxic evaluation of some novel hexahydroquinoline derivatives containing benzofuran moiety. Res. Chem. Intermed. 2016, 42, 1863–1883. [Google Scholar] [CrossRef]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R.U.S. FDA Approved Drugs from 2015–June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef]
- Abdelaziz, M.E.; El-Miligy, M.M.M.; Fahmy, S.M.; Mahran, M.A.; Hazzaa, A.A. Design, synthesis and docking study of pyridine and thieno[2,3-b] pyridine derivatives anticancer PIM-1 kinase inhibitors. Bioorg. Chem. 2018, 80, 674–692. [Google Scholar] [CrossRef]
- Eurtivong, C.; Semenov, V.; Semenova, M.; Konyushkin, L.; Atamanenko, O.; Reynisson, J.; Kiselyov, A. 3-Amino-thieno[2,3-b]pyridines as microtubule-destabilising agents: Molecular modelling and biological evaluation in the sea urchin embryo and human cancer cells. Bioorg. Med. Chem. 2017, 25, 658–664. [Google Scholar] [CrossRef]
- Al-Trawneh, S.A.; Tarawneh, A.H.; Gadetskaya, A.V.; Seo, E.; Al-Ta’ani, M.R.; Al-Taweel, S.A.; El-Abadelah, M.M. Synthesis and cytotoxicity of thieno[2,3-b]pyridine derivatives toward sensitive and multidrug-resistant leukemia cells. Acta Chim. Solv. 2021, 68, 458–465. [Google Scholar] [CrossRef]
- Elsherif, M.A. Antibacterial evaluation and molecular properties of pyrazolo[3,4-b]pyridines and thieno[2,3-b] pyridines. J. Appl. Pharm. Sci. 2021, 11, 118–124. [Google Scholar] [CrossRef]
- Mohi El-Deen, E.M.; Abd El-Meguid, E.A.; Hasabelnaby, S.; Karam, E.A.; Nossier, E.S. Synthesis, Docking Studies, and In Vitro Evaluation of Some Novel Thienopyridines and Fused Thienopyridine–Quinolines as Antibacterial Agents and DNA Gyrase Inhibitors. Molecules 2019, 24, 3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekky, A.E.M.; Sanad, S.M.H.; Said, A.Y.; Elneairy, M.A.A. Synthesis, cytotoxicity, in-vitro antibacterial screening and in-silico study of novel thieno[2,3-b]pyridines as potential pim-1 inhibitors. Synth. Commun. 2020, 50, 2376–2389. [Google Scholar] [CrossRef]
- Attaby, F.A.; Abdel-Fattah, A.M.; Shaif, L.M.; Elsayed, M.M. Reactions, Anti-Alzheimer and Anti COX-2 Activities of the Newly Synthesized 2-Substituted Thienopyridines. Curr. Org. Chem. 2009, 13, 1654–1663. [Google Scholar] [CrossRef]
- Binsaleh, N.K.; Wigley, C.A.; Whitehead, K.A.; van Rensburg, M.; Reynisson, J.; Pilkington, L.I.; Barker, D.; Jones, S.; Dempsey-Hibbert, N.C. Thieno[2,3-b]pyridine derivatives are potent anti-platelet drugs, inhibiting platelet activation, aggregation and showing synergy with aspirin. Eur. J. Med. Chem. 2018, 143, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Amorim, R.; de Meneses, M.D.F.; Borges, J.C.; da Silva Pinheiro, L.C.; Caldas, L.A.; Cirne-Santos, C.C.; de Mello, M.V.P.; de Souza, A.M.T.; Castro, H.C.; de Palmer Paixão, I.C.N.; et al. Thieno[2,3-b]pyridine derivatives: A new class of antiviral drugs against Mayaro virus. Arch. Virol. 2017, 162, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.Y.; Zuo, W.Q.; Xu, Y.; Gao, C.; Zeng, X.X.; Zhang, L.D.; You, X.Y.; Peng, C.T.; Shen, Y.; Yang, S.Y.; et al. Discovery and structure−activity relationships study of novel thieno[2,3-b]pyridine analogues as hepatitis C virus inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Y.; Wang, X.Y.; Wang, B.; He, H.Y.; Liu, J.Y.; Xiang, M.L.; He, J.; Wu, X.H.; Yang, L. Synthesis, preliminary structure-activity relationships, and in vitro biological evaluation of 6-aryl-3-amino-thieno[2,3-b]pyridine derivatives as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2013, 23, 2349–2352. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, R.B.S. Biological Activity of Pyrimidine Derivativies: A Review. Org. Med. Chem. IJ. 2017, 2, 555581. [Google Scholar] [CrossRef]
- Patil, S.B. Biological and medicinal significance of pyrimidines: A review. Int. J. Pharm Sci. Res. 2018, 9, 44–52. [Google Scholar] [CrossRef]
- Kaur, H.; Machado, M.; de Kock, C.; Smith, P.; Chibale, K.; Prudêncio, M.; Singh, K. Primaquine-pyrimidine hybrids: Synthesis and dual-stage antiplasmodial activity. Eur. J. Med. Chem. 2015, 101, 266–273. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, S.M.; Al-Majid, A.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.K.; Yousuf, S.; Choudhary, M.I.; Wadood, A. Synthesis and structure investigation of novel pyrimidine-2,4,6-trione derivatives of highly potential biological activity as anti-diabetic agent. J. Mol. Struct. 2015, 1098, 365–376. [Google Scholar] [CrossRef]
- Su, L.; Li, J.; Zhou, Z.; Huang, D.; Zhang, Y.; Pei, H.; Guo, W.; Wu, H.; Wang, X.; Liu, M.; et al. Corrigendum to Design, synthesis and evaluation of hybrid of tetrahydrocarbazole with 2,4-diaminopyrimidine scaffold as antibacterial agents. Eur. J. Med. Chem. 2019, 168, 385. [Google Scholar] [CrossRef] [PubMed]
- Bassyouni, F.; Tarek, M.; Salama, A.; Ibrahim, B.; Salah El Dine, S.; Yassin, N.; Hassanein, A.; Moharam, M.; Abdel-Rehim, M. Promising Antidiabetic and Antimicrobial Agents Based on Fused Pyrimidine Derivatives: Molecular Modeling and Biological Evaluation with Histopathological Effect. Molecules 2021, 26, 2370. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Pyrimidine-based EGFR TK Inhibitors in Targeted Cancer Therapy. Eur. J. Med. Chem. 2021, 221, 113523. [Google Scholar] [CrossRef]
- Tylińska, B.; Wiatrak, B.; Czyżnikowska, Ż.; Cieśla-Niechwiadowicz, A.; Gębarowska, E.; Janicka-Kłos, A. Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 3825. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.; Prasad, T.; Sharma, T. Pyrimidine: A review on anticancer activity with key emphasis on SAR. Futur. J. Pharm. Sci. 2021, 7, 123. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, M.; Park, M.; Tae, J.; Baek, D.J.; Park, K.D.; Choo, H. Synthesis of novel dihydropyridothienopyrimidin-4, 9-dione derivatives. Molecules 2015, 20, 5074–5084. [Google Scholar] [CrossRef] [Green Version]
- Mohi El-Deen, E.M.; Abd El-Meguid, E.A.; Karam, E.A.; Nossier, E.S.; Ahmed, M.F. Synthesis and Biological Evaluation of New Pyridothienopyrimidine Derivatives as Antibacterial Agents and Escherichia coli Topoisomerase II Inhibitors. Antibiotics 2020, 9, 695. [Google Scholar] [CrossRef]
- Sanad, S.M.H.; Mekky, A.E.M. New pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(3H)-one hybrids linked to arene units: Synthesis of potential MRSA, VRE, and COX-2 inhibitors. Can. J. Chem. 2021, 99, 900–909. [Google Scholar] [CrossRef]
- Sirakanyan, S.N.; Spinelli, D.; Geronikaki, A.; Hakobyan, E.K.; Sahakyan, H.; Arabyan, E.; Zakaryan, H.; Nersesyan, L.E.; Aharonyan, A.S.; Danielyan, I.S.; et al. Synthesis, Antitumor Activity, and Docking Analysis of New Pyrido[3′,2′:4,5]furo(thieno)[3,2-d]pyrimidin-8-amines. Molecules 2019, 24, 3952. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.A.; Kim, M.; Kim, J.Y.; Shin, Y.; Song, J.; Jeong, J. A novel pyrido-thieno-pyrimidine derivative activates p53 through induction of phosphorylation and acetylation in colorectal cancer cells. Int. J. Oncol. 2015, 46, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, Y.M.A.; Said, M.M.; El Shihawy, H.A.; Abouzid, K.A. Discovery of novel tricyclic pyrido [3′, 2′: 4, 5] thieno [3, 2-d] pyrimidin-4-amine derivatives as VEGFR-2 inhibitors. Bioorg. Chem. 2015, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Deau, E.; Marchand, P.; Nourrisson, M.-R.; Logé, C.; Coadou, G.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and molecular modelling studies of 8-arylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015, 92, 124. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Bushra Begum, A.; Zabiulla, S.; Mamatha, S.V.; Ara Khanum, S. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. J. Chem. Pharm. Res. 2015, 7, 281–301. [Google Scholar] [CrossRef]
- Sepsey Für, C.; Riszter, G.; SzigetvárI, Á.; Dékány, M.; Keglevich, G.; HazaI, L.; BölcskeI, H. Novel Ring Systems: Spiro[Cycloalkane] Derivatives of Triazolo- and Tetrazolo-Pyridazines. Molecules 2021, 26, 2140. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Suresh, B.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorganic Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- Delost, M.D.; Smith, D.T.; Anderson, B.J.; Njardarson, J.T. From Oxiranes to Oligomers: Architectures of U.S. FDA Approved Pharmaceuticals Containing Oxygen Heterocycles. J. Med. Chem. 2018, 61, 10996–11020. [Google Scholar] [CrossRef]
- Chauhan, R.; Saini, P.; Choudhary, R.; Rani, S. A Review on Pharmacological Profile of Ethanamide and their Derivatives. Int. J. Pharm. Sci. Rev. Res. 2020, 64, 162–170. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- de Castro Barbosa, M.L.; Lima, L.M.; Tesch, R.; Sant’ Anna, C.M.R.; Totzke, F.; Kubbutat, M.H.; Schächtele, C.; Laufer, S.; Barreiro, E.J. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2014, 71, 1–14. [Google Scholar] [CrossRef]
- Elshaier, Y.A.; Shaaban, M.A.; Abd El Hamid, M.K.; Abdelrahman, M.H.; Abou-Salim, M.A.; Elgazwi, S.M.; Halaweish, F. Design and synthesis of pyrazolo [3, 4-d] pyrimidines: Nitric oxide releasing compounds targeting hepatocellular carcinoma. Bioorg. Med. Chem. 2017, 25, 2956–2970. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Ren, H.; Zhao, M.; Chong, Y.; Zhao, W.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef] [PubMed]
- van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Aiebchun, T.; Mahalapbutr, P.; Auepattanapong, A.; Khaikate, O.; Seetaha, S.; Tabtimmai, L.; Kuhakarn, C.; Choowongkomon, K.; Rungrotmongkol, T. Identification of vinyl sulfone derivatives as egfr tyrosine kinase inhibitor: In vitro and in silico studies. Molecules 2021, 26, 2211. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Erlotinib: A pharmacoeconomic review of its use in advanced non-small cell lung cancer. Pharmacoeconomics 2010, 28, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Youssefyeh, R.D.; Brown, R.E.; Wilson, J.; Shah, U.; Jones, H.; Loev, B.; Khandwala, A.; Leibowitz, M.J.; Sonnino-Goldman, P. Pyrido [3′, 2′: 4, 5] thieno [3, 2-d]-N-triazines: A new series of orally active antiallergic agents. J. Med. Chem. 1984, 27, 1639–1643. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Mizutani, M.Y.; Takamatsu, Y.; Ichinose, T.; Nakamura, K.; Itai, A. Effective handling of induced-fit motion in flexible docking. Proteins Struct. Funct. Bioinform. 2006, 63, 878–891. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. No. | Gram +ve Bacteria | Gram −ve Bacteria | |||
|---|---|---|---|---|---|
| S. aureus | B. subtilis | B. cereus | E. coli | S. typhimurium | |
| 2a | NA | NA | 64 | NA | NA |
| 2b | 64 | 128 | 64 | 128 | 64 |
| 3a | 4 | 4 | 8 | 8 | 16 |
| 3b | 32 | 8 | 32 | 16 | 32 |
| 4a | 4 | 8 | 4 | 8 | 16 |
| 4b | 8 | 128 | 16 | 128 | 128 |
| 5a | 8 | 8 | 16 | 8 | 16 |
| 5b | 8 | 8 | 16 | 32 | 32 |
| 6a | 32 | 16 | 8 | 32 | 16 |
| 6b | 4 | 8 | 16 | 8 | 16 |
| 6c | 4 | 4 | 8 | 8 | 16 |
| 6d | 8 | 32 | 8 | 32 | 32 |
| 7a | 4 | NA | 8 | 8 | 16 |
| 7b | 64 | 64 | 64 | 64 | 128 |
| 7c | 32 | 32 | 8 | 32 | 32 |
| 7d | 4 | 64 | 64 | 32 | 32 |
| 8a | 8 | 16 | 8 | 32 | 32 |
| 8b | 4 | 8 | 16 | 8 | 16 |
| 9a | 16 | 32 | 8 | 32 | 16 |
| 9b | 4 | 4 | 8 | 4 | 16 |
| Amoxicillin | 4 | 8 | 16 | 8 | 16 |
| Compd. No. | Yeasts | Fungi | |||
|---|---|---|---|---|---|
| C. albicans | C. tropicals | S. cerevisiae | A. flavus | A. niger | |
| 2a | 128 | 128 | 16 | NA | NA |
| 2b | 64 | 32 | 32 | 32 | 32 |
| 3a | 16 | 8 | 8 | 8 | 8 |
| 3b | 32 | 16 | 64 | 32 | 16 |
| 4a | 16 | 16 | 8 | 8 | 8 |
| 4b | 8 | 16 | 8 | 16 | 16 |
| 5a | 16 | 8 | 8 | 8 | 16 |
| 5b | 64 | 16 | 16 | 32 | 64 |
| 6a | 64 | 16 | 32 | 64 | 32 |
| 6b | 8 | 8 | 8 | 16 | 8 |
| 6c | 16 | 4 | 16 | 4 | 8 |
| 6d | 64 | 64 | 32 | 32 | 16 |
| 7a | 16 | 16 | 8 | 16 | 16 |
| 7b | 64 | 64 | 32 | 64 | 128 |
| 7c | 16 | 8 | 8 | 4 | 16 |
| 7d | 32 | 64 | 32 | 64 | 64 |
| 8a | 64 | 16 | 16 | 16 | 16 |
| 8b | 8 | 16 | 16 | 8 | 8 |
| 9a | 16 | 32 | 32 | 16 | 64 |
| 9b | 4 | 4 | 8 | 8 | 8 |
| Clotrimazole | 16 | 8 | 8 | 8 | 8 |
| Compd. No. | HepG2 (µM) | MCF7 (µM) | WISH |
|---|---|---|---|
| 2a | 56.57 ± 3.64 | 64.34 ± 2.91 | |
| 2b | 33.21 ± 1.79 | 42.39 ± 2.24 | |
| 3a | 2.31 ± 0.35 | 7.24 ± 0.64 | 416.83 ± 15.17 |
| 3b | 11.34 ± 0.65 | 24.72 ± 1.32 | |
| 4a | 2.99 ± 0.15 | 15.42 ± 0.45 | 460.23 ± 11.08 |
| 4b | 36.52 ± 1.82 | 43.27 ± 2.28 | |
| 5a | 1.99 ± 0.09 | 2.79 ± 0.18 | 408.48 ± 15.93 |
| 5b | 10.16 ± 0.29 | 21.06 ± 1.16 | |
| 6a | 52.18 ± 1.45 | 77.41 ± 3.62 | |
| 6b | 2.75 ± 0.13 | 9.89 ± 0.55 | 394.98 ± 10.20 |
| 6c | 12.11 ± 0.33 | 22.24 ± 0.67 | |
| 6d | 4.45 ± 0.22 | 21.67 ± 0.70 | |
| 7a | 26.05 ± 1.89 | 28.11 ± 0.92 | |
| 7b | 39.74 ± 1.89 | 41.62 ± 2.20 | |
| 7c | 10.35 ± 0.31 | 20.9 ± 0.93 | |
| 7d | 23.25 ± 0.35 | 26.55 ± 1.62 | |
| 8a | 6.78 ± 0.73 | 20.88 ± 1.46 | |
| 8b | 2.79 ± 0.08 | 13.54 ± 0.76 | 401.37± 17.32 |
| 9a | 4.88 ± 0.65 | 23.56 ± 1.24 | |
| 9b | 1.17 ± 0.09 | 1.52 ± 0.08 | 417.55 ± 14.1 |
| Doxorubicin | 2.85 ± 0.21 | 3.58 ± 0.33 | 432.10 ± 19.30 |
| Compound No. | EGFR IC50 (nM) |
|---|---|
| 3a | 17.29 ± 0.24 |
| 4a | 53.57 ± 0.41 |
| 5a | 9.66 ± 0.08 |
| 6b | 53.19 ± 0.46 |
| 8b | 38.44 ± 0.25 |
| 9b | 7.27 ± 0.11 |
| Erlotinib | 27.01 ± 0.16 |
| Compound | S (kcal/mol) | Amino Acids | Interacting Groups | Type of Bond | Length (Å) |
|---|---|---|---|---|---|
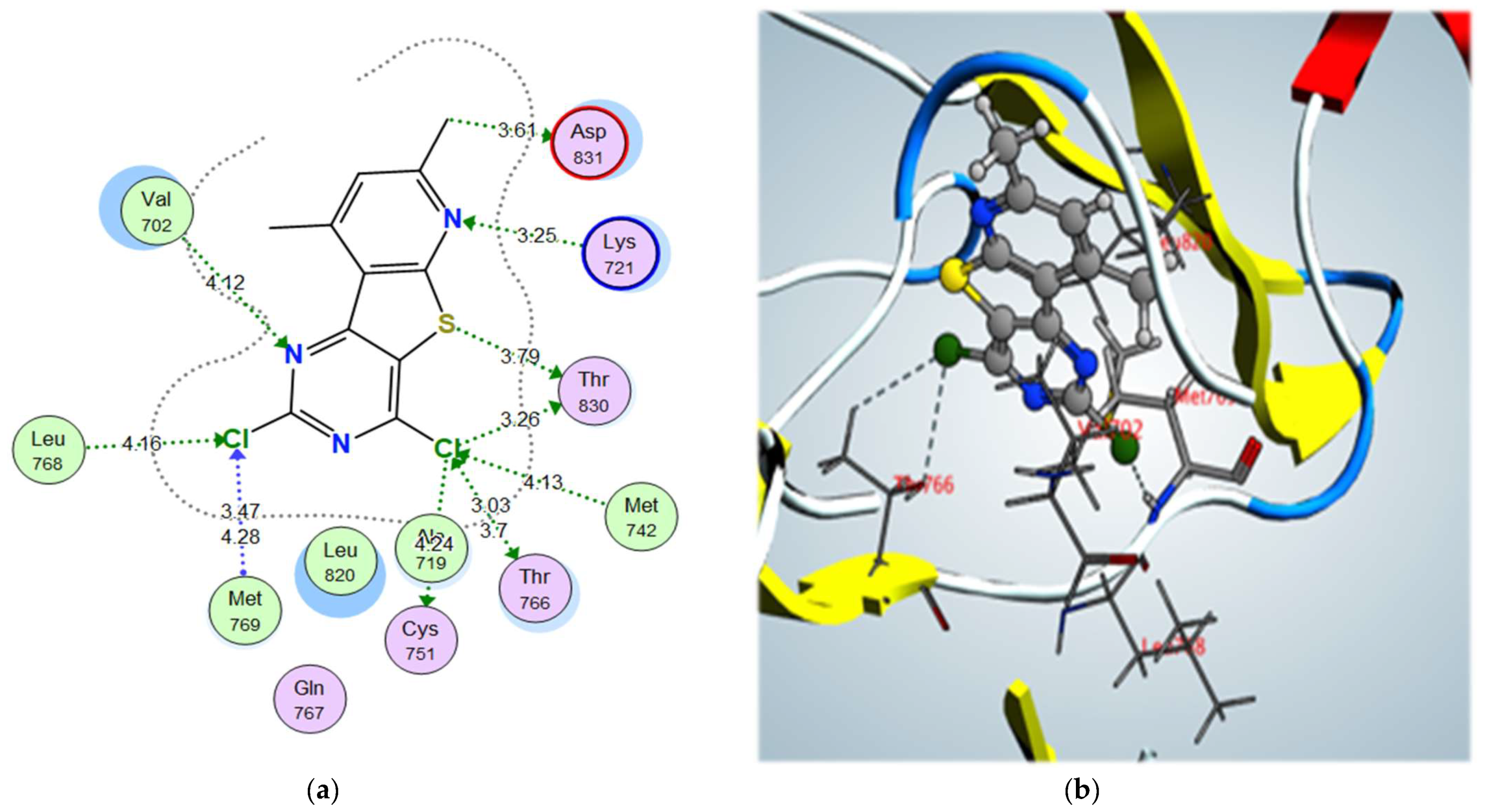
| 3a | –11.42 | Val702 | N (Pyrimidine) | H-bond acceptor | 4.12 |
| Lys721 | N (Pyridine) | H-bond acceptor | 3.25 | ||
| Met769 | Cl (Pyrimidine) | Halogen bond | 3.47 | ||
| Leu768 | Cl (Pyrimidine) | Halogen bond | 4.16 | ||
| Thr830 | Cl (Pyridine) | Halogen bond | 3.26 | ||
| Met742 | Cl (Pyridine) | Halogen bond | 4.13 | ||
| Thr766 | Cl (Pyridine) | Halogen bond | 3.03 | ||
| Thr830 | S | σ-hole bond | 3.79 | ||
| 4a | –8.94 | Lys721 | Cl | Halogen bond | 3.69 |
| Cys751 | O (C=O) | σ-hole bond | 3.78 | ||
| Thr766 | S | σ-hole bond | 4.21 | ||
| Met769 | S and N (Pyridine) | H-bond acceptor | 3.41/3.55 | ||
| Leu768 | N (Pyridine) | H-bond acceptor | 3.84 | ||
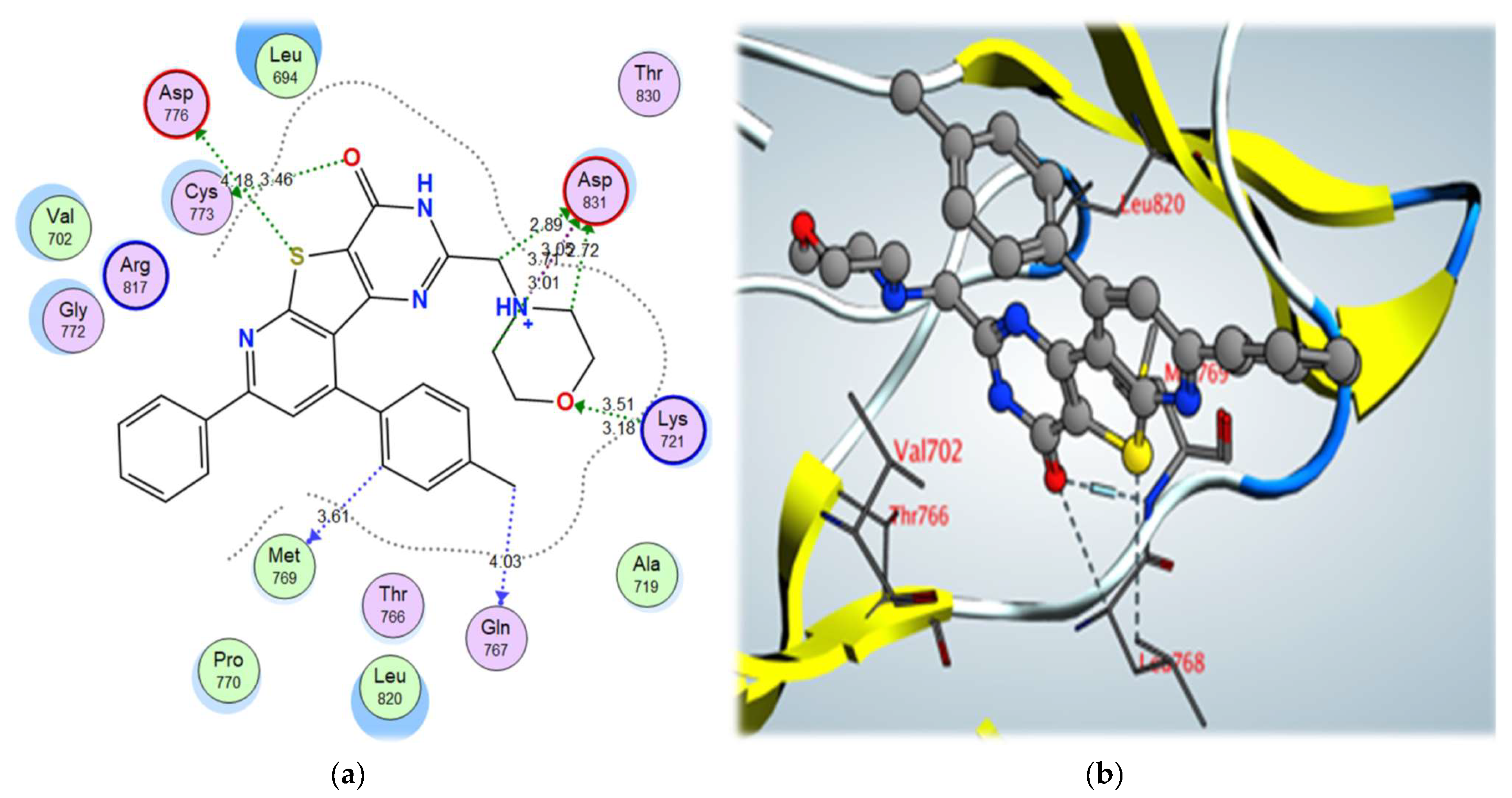
| 5a | –11.48 | Asp831 | NH+ | Ionic interaction | 3.71 |
| Lys721 | O (Morpholine) | H-bond acceptor | 3.51 | ||
| Cys773 | O (C=O) | σ-hole bond | 3.46 | ||
| Asp776 | S | σ-hole bond | 4.18 | ||
| 6b | –10.06 | Leu820 | NH | H-bond acceptor | 3.78 |
| Cys751 | O (C=O) | H-bond acceptor | 3.49 | ||
| Gln767 | S | σ-hole bond | 3.12 | ||
| Thr766 | S | σ-hole bond | 3.87 | ||
| Met769 | S | H-bond acceptor | 3.83 | ||
| 8b | –9.11 | Leu694 | S (Thiophene) | H-bond acceptor | 4.38 |
| Leu694 | S (Side chain) | σ-hole bond | 3.79 | ||
| Thr766 | N (Pyridine) | H-bond acceptor | 3.71 | ||
| 9b | –12.01 | Met769 | S (thiophene) | H-bond acceptor | 4.09 |
| Leu768 | S (thiophene) | H-bond acceptor | 4.37 | ||
| Leu820 | S (thiophene) | H-bond acceptor | 4.47 | ||
| Thr766 | S (Side chain) | σ-hole bond | 4.20 | ||
| Leu820 | N and NH (Pyrimidine) | H-bond acceptor | 3.73/3.74 | ||
| Val702 | O (Oxirane) | H-bond acceptor | 3.64 | ||
| erlotinib | –10.48 | Leu768 | N (Pyrimidine) | H-bond acceptor | 3.64 |
| Met769 | N (Pyrimidine) | H-bond acceptor | 2.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohi El-Deen, E.M.; Anwar, M.M.; El-Gwaad, A.A.A.; Karam, E.A.; El-Ashrey, M.K.; Kassab, R.R. Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents. Molecules 2022, 27, 803. https://doi.org/10.3390/molecules27030803
Mohi El-Deen EM, Anwar MM, El-Gwaad AAA, Karam EA, El-Ashrey MK, Kassab RR. Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents. Molecules. 2022; 27(3):803. https://doi.org/10.3390/molecules27030803
Chicago/Turabian StyleMohi El-Deen, Eman M., Manal M. Anwar, Amina A. Abd El-Gwaad, Eman A. Karam, Mohamed K. El-Ashrey, and Rafika R. Kassab. 2022. "Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents" Molecules 27, no. 3: 803. https://doi.org/10.3390/molecules27030803
APA StyleMohi El-Deen, E. M., Anwar, M. M., El-Gwaad, A. A. A., Karam, E. A., El-Ashrey, M. K., & Kassab, R. R. (2022). Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents. Molecules, 27(3), 803. https://doi.org/10.3390/molecules27030803