
Hydrophobic Eutectic Solvent-Based Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Pesticides in Wine



Abstract
:1. Introduction
2. Results and Discussion
2.1. Fine-Tuning of the Extraction Procedure
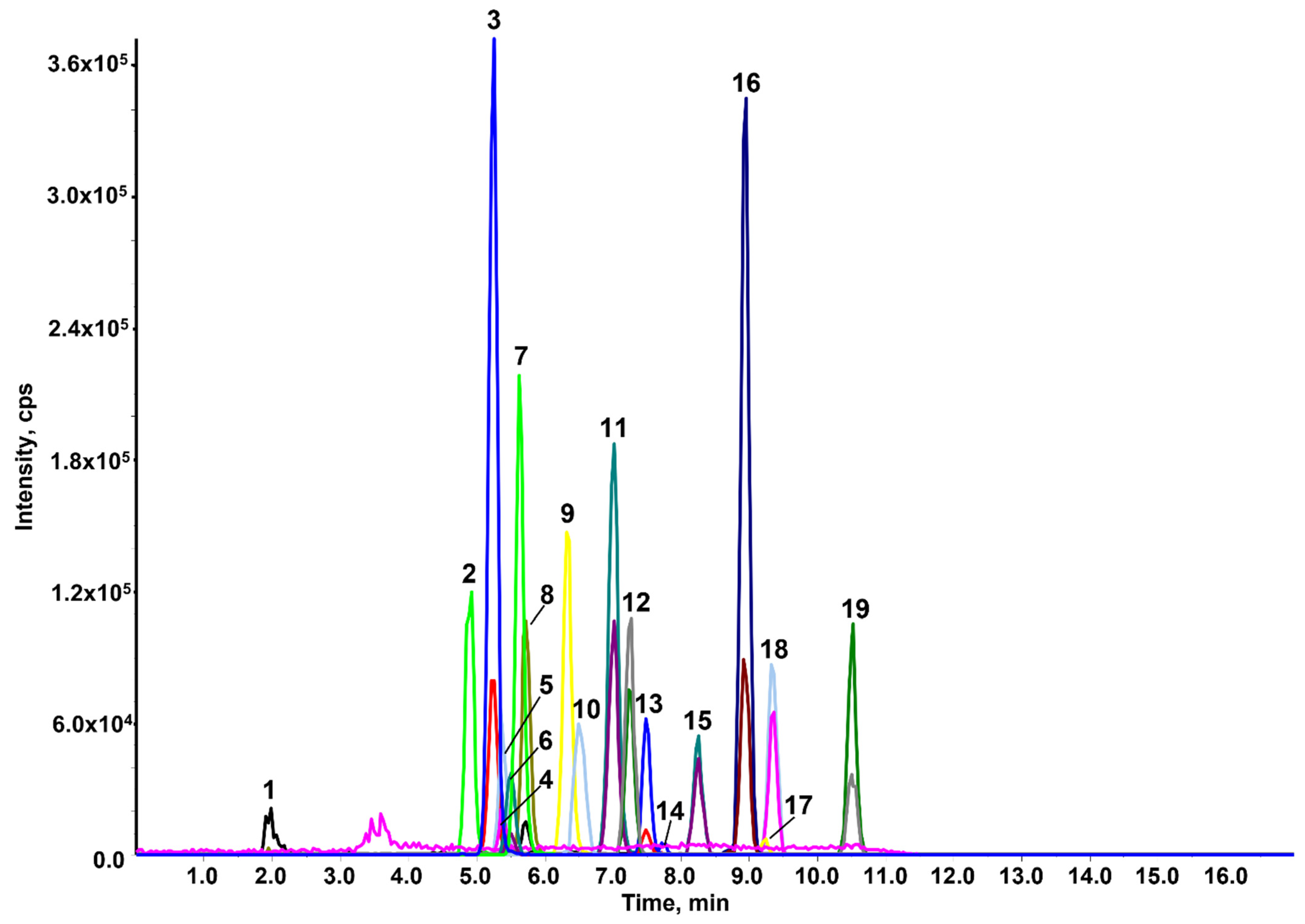
2.2. Fine-Tuning of the Chromatographic Conditions
2.3. Validation Results
2.4. Comparison with Other Methods
2.5. Results on Real Samples
3. Materials and Methods
3.1. Chemicals, Materials and Solutions
3.2. Wine Samples
3.3. Preparation of the Eutectic Mixture and Extraction Procedure
3.4. HPLC-MS Analysis
3.5. Method Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cabras, P.; Angioni, A. Pesticide residues in grapes, wine and their processing products. J. Agric. Food Chem. 2000, 48, 967–973. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzo, G.; Gentili, A.; Marchese, S.; Perret, D. Development of a method based on liquid chromatography–electrospray mass spectrometry for analyzing imidazolinone herbicides in environmental water at part-per-trillion levels. J. Chromatogr. A 1998, 800, 109–119. [Google Scholar] [CrossRef]
- Marchese, S.; Perret, D.; Gentili, A.; D’Ascenzo, G.; Faberi, A. Determination of phenoxyacid herbicides and their phenolic metabolites in surface and drinking water. Rapid Commun. Mass Spectrom. 2002, 16, 134–141. [Google Scholar] [CrossRef]
- D’Ascenzo, G.; Gentili, A.; Marchese, S.; Marino, A.; Perret, D. Simultaneous determination of base/neutral and acid herbicides in natural water at the part per trillion level. Chromatographia 1998, 48, 497–505. [Google Scholar] [CrossRef]
- European Union. Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending council directive 91/414/EEC 2020 (consolidated text). Off. J. Eur. Union 2005, 70, 1–16. [Google Scholar]
- Narenderan, S.T.; Meyyanathan, S.N.; Babu, B. Review of pesticide residue analysis in fruits and vegetables. Pre-treatment, extraction and detection techniques. Food Res. Int. 2020, 133, 109141. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “Dispersive Solid-Phase Extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–443. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Jiao, B. Determination of ten pyrethroids in various fruit juices: Comparison of dispersive liquid-liquid microextraction sample preparation and QuEChERS method combined with dispersive liquid-liquid microextraction. Food Chem. 2014, 159, 367–373. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Hosseini, M.R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Yang, P.; Li, H.; Wang, H.; Han, F.; Jing, S.; Yuan, C.; Guo, A.; Zhang, Y.; Xu, Z. Dispersive liquid-liquid microextraction method for HPLC determination of phenolic compounds in wine. Food Anal. Methods 2017, 10, 2383–2397. [Google Scholar] [CrossRef]
- Santana-Mayor, Á.; Rodríguez-Ramos, R.; Socas-Rodríguez, B.; Rodríguez-Delgado, M.Á.; D’Orazio, G. Nano-liquid chromatography combined with a sustainable microextraction based on natural deep eutectic solvents for analysis of phthalate esters. Electrophoresis 2020, 41, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Campone, L.; Piccinelli, A.L.; Rastrelli, L. Dispersive liquid–liquid microextraction combined with high-performance liquid chromatography–tandem mass spectrometry for the identification and the accurate quantification by isotope dilution assay of Ochratoxin A in wine samples. Anal. Bioanal. Chem. 2011, 399, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Hrouzková, S.; Brišová, M.; Szarka, A. Development of fast, efficient and ecological method employing vortex-assisted dispersive liquid–liquid microextraction combined with fast gas chromatography–mass spectrometry for pesticide residues analysis in alcohol-content samples. J. Chromatogr. A 2017, 1506, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cabo, T.; Rodríguez, I.; Ramil, M.; Cela, R. Dispersive liquid—Liquid microextraction using non-chlorinated, lighter than water solvents for gas chromatography–mass spectrometry determination of fungicides in wine. J. Chromatogr. A 2011, 1218, 6603–6611. [Google Scholar] [CrossRef]
- Chu, S.P.; Tseng, W.C.; Kong, P.H.; Huang, C.K.; Chen, J.H.; Chen, P.S.; Huang, S.D. Up-and-down-shaker-assisted dispersive liquid—Liquid microextraction coupled with gas chromatography–mass spectrometry for the determination of fungicides in wine. Food Chem. 2015, 185, 377–382. [Google Scholar] [CrossRef]
- Carbonell-Rozas, L.; Canales, R.; Lara, F.J.; García-Campaña, A.M.; Silva, M.F. A natural deep eutectic solvent as a novel dispersive solvent in dispersive liquid—Liquid microextraction based on solidification of floating organic droplet for the determination of pesticide residues. Anal. Bioanal. Chem. 2021, 413, 6413–6424. [Google Scholar] [CrossRef]
- Jia, L.; Huang, X.; Zhao, W.; Wang, H.; Jing, X. An effervescence tablet-assisted microextraction based on the solidification of deep eutectic solvents for the determination of strobilurin fungicides in water, juice, wine, and vinegar samples by HPLC. Food Chem. 2020, 317, 126424. [Google Scholar] [CrossRef]
- Timofeeva, I.; Kanashina, D.; Moskvin, L.; Bulatov, A. An evaporation-assisted dispersive liquid–liquid microextraction technique as a simple tool for high performance liquid chromatography tandem–mass spectrometry determination of insecticides in wine. J. Chromatogr. A 2017, 1512, 107–114. [Google Scholar] [CrossRef]
- Chen, B.; Wu, F.Q.; Wu, W.D.; Jin, B.H.; Xie, L.Q.; Feng, W.; Ouyang, G. Determination of 27 pesticides in wine by dispersive liquid—Liquid microextraction and gas chromatography–mass spectrometry. Microchem. J. 2016, 126, 415–422. [Google Scholar] [CrossRef]
- Cinelli, G.; Avino, P.; Notardonato, I.; Russo, M.V. Ultrasound-vortex-assisted dispersive liquid—Liquid microextraction coupled with gas chromatography with a nitrogen–phosphorus detector for simultaneous and rapid determination of organophosphorus pesticides and triazines in wine. Anal. Methods 2014, 6, 782–790. [Google Scholar] [CrossRef]
- Zacharis, C.K.; Christophoridis, C.; Fytianos, K. Vortex-assisted liquid–liquid microextraction combined with gas chromatography-mass spectrometry for the determination of organophosphate pesticides in environmental water samples and wines. J. Sep. Sci. 2012, 35, 2422–2429. [Google Scholar] [CrossRef]
- D’Orazio, G.; Gentili, A.; Fanali, S.; Fanali, C.; Dal Bosco, C. Innovative solutions for the extraction of vitamins from pharmaceutical and biological samples. Curr. Anal. Chem. 2021, 17, 1114–1132. [Google Scholar] [CrossRef]
- Li, G.; Row, K.H. Utilization of deep eutectic solvents in dispersive liquid-liquid micro-extraction. TrAC Trends Anal. Chem. 2019, 120, 115651. [Google Scholar] [CrossRef]
- Gallo, V.; Tomai, P.; Di Lisio, V.; Dal Bosco, C.; D’Angelo, P.; Fanali, C.; D’Orazio, G.; Silvestro, I.; Picó, Y.; Gentili, A. Application of a low transition temperature mixture for the dispersive liquid—Liquid microextraction of illicit drugs from urine samples. Molecules 2021, 26, 5222. [Google Scholar] [CrossRef] [PubMed]
- Tomai, P.; Lippiello, A.; D’Angelo, P.; Persson, I.; Martinelli, A.; Di Lisio, V.; Curini, R.; Fanali, C.; Gentili, A. A low transition temperature mixture for the dispersive liquid-liquid microextraction of pesticides from surface waters. J. Chromatogr. A 2019, 1605, 360329. [Google Scholar] [CrossRef]
- Rykowska, I.; Ziemblińska, J.; Nowak, I. Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: A review. J. Mol. Liq. 2018, 259, 319–339. [Google Scholar] [CrossRef]
- Dal Bosco, C.; Di Lisio, V.; D’Angelo, P.; Gentili, A. Hydrophobic eutectic solvent with antioxidant properties: Application for the dispersive liquid—Liquid microextraction of fat-soluble micronutrients from fruit juices. ACS Sustain. Chem. Eng. 2021, 24, 8170–8178. [Google Scholar] [CrossRef]
- Gallo, V.; Tomai, P.; Gherardi, M.; Fanali, C.; De Gara, L.; D’Orazio, G.; Gentili, A. Dispersive liquid-liquid microextraction using a low transition temperature mixture and liquid chromatography-mass spectrometry analysis of pesticides in urine samples. J. Chromatogr. A 2021, 1642, 462036. [Google Scholar] [CrossRef]
- Cortese, M.; Gigliobianco, M.R.; Magnoni, F.; Censi, R.; Di Martino, P. Compensate for or minimize matrix effects? Strategies for overcoming matrix effects in liquid chromatography-mass spectrometry technique: A tutorial review. Molecules 2020, 25, 3047. [Google Scholar] [CrossRef]
- WHO. Pesticide residues in food—Report 2003. In Proceedings of the Joint Meeting of the FAO Panel of Experts on Pesticide Residues in Food and the Environment and the WHO Core Assessment Group on Pesticide Residues, Geneva, Switzerland, 15–24 September 2003. [Google Scholar]
- Jiang, Y.; Li, X.; Xu, J.; Pan, C.; Zhang, J.; Niu, W. Multiresidue method for the determination of 77 pesticides in wine using QuEChERS sample preparation and gas chromatography with mass spectrometry. Food Addit. Contam. 2009, 26, 859–866. [Google Scholar] [CrossRef]
- Hill, H.M. Bioanalytical methods validation: A critique of the proposed FDA guidance. Chromatographia 2000, 52, S65–S69. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Analyte a | Regression Equation (n = 8) | R2 b | Matrix Effect % | |
|---|---|---|---|---|
| b ± Sb·t(0.05;6) | a ± Sa·t(0.05;6) | |||
| AZX | −7.3 | |||
| Solvent | 206.10 ± 10.70 | 25.70 ± 1.40 | 0.9894 | |
| Matrix | 191.40 ± 12.70 | 10.20 ± 0.70 | 0.9992 | |
| BSC | −19 | |||
| Solvent | 36.40 ± 5.30 | 2.67 ± 0.29 | 0.9931 | |
| Matrix | 29.50 ± 2.90 | 0.42 ± 0.05 | 0.9964 | |
| BPR | 21 | |||
| Solvent | 9.79 ± 1.23 | 4.63 ± 0.55 | 0.9583 | |
| Matrix | 11.80 ± 0.89 | 7.98 ± 0.88 | 0.8908 | |
| CPS | 14 | |||
| Solvent | 2.98 ± 0.45 | - | 0.9989 | |
| Matrix | 3.11 ± 0.47 | - | 0.9992 | |
| CPM | 18 | |||
| Solvent | 2.79 ± 0.39 | 0.21 ±0.03 | 0.977 | |
| Matrix | 3.29 ± 0.48 | −0.52 ± 0.08 | 0.9673 | |
| CLF | −31 | |||
| Solvent | 33.50 ± 4.20 | 4.35 ± 0.66 | 0.9993 | |
| Matrix | 23.20 ± 3.40 | 8.88 ± 1.29 | 0.9712 | |
| DOD | −2.8 | |||
| Solvent | 21.50 ± 1.10 | 1.09 ±0.05 | 0.9982 | |
| Matrix | 20.90 ± 0.90 | 1.89 ± 0.08 | 0.9972 | |
| FLD | −20 | |||
| Solvent | 0.35 ± 0.04 | - | 0.9375 | |
| Matrix | 0.28 ± 0.03 | - | 0.9693 | |
| HXT | −22 | |||
| Solvent | 47.80 ± 3.10 | −2.94 ± 0.17 | 0.9994 | |
| Matrix | 37.10 ± 2.60 | −14.30 ± 0.80 | 0.9779 | |
| MXF | −15 | |||
| Solvent | 165.50 ± 14.90 | 36.50 ± 3.30 | 0.9885 | |
| Matrix | 141.00 ± 11.30 | 4.45 ± 0.41 | 0.9993 | |
| MYC | −13 | |||
| Solvent | 47.20 ± 2.40 | 0.14 ± 0.01 | 0.9924 | |
| Matrix | 40.90 ± 2.50 | −8.90 ± 0.50 | 0.9921 | |
| PEN | 1.7 | |||
| Solvent | 12.10 ± 1.30 | 12.30 ± 1.40 | 0.9058 | |
| Matrix | 12.30 ± 1.50 | −6.37 ± 0.77 | 0.8661 | |
| PYR | −12 | |||
| Solvent | 64.10 ± 8.30 | 0.93 ± 0.12 | 0.995 | |
| Matrix | 56.50 ± 7.30 | −13.50 ± 1.70 | 0.9923 | |
| PRD | −26 | |||
| Solvent | 115.10 ± 16.30 | −2.90 ± 0.42 | 0.9971 | |
| Matrix | 84.90 ± 12.40 | −21.50 ± 3.10 | 0.9912 | |
| PPF | 18 | |||
| Solvent | 84.40 ± 12.70 | −2.88 ± 0.43 | 0.9869 | |
| Matrix | 100.20 ± 15.20 | 27.40 ± 4.10 | 0.9961 | |
| PRO | 18 | |||
| Solvent | 46.00 ± 5.80 | −8.38 ±1.28 | 0.9938 | |
| Matrix | 37.70 ± 4.90 | 6.72 ± 1.04 | 0.9803 | |
| STM | −15 | |||
| Solvent | 58.30 ± 5.80 | −27.40 ± 3.01 | 0.957 | |
| Matrix | 49.60 ± 4.80 | −38.50 ± 4.20 | 0.9299 | |
| TEB | −15 | |||
| Solvent | 88.20 ± 7.10 | 0.73 ± 0.06 | 0.9958 | |
| Matrix | 74.80 ± 5.80 | −11.00 ± 0.80 | 0.9978 | |
| TBF | −22 | |||
| Solvent | 23.30 ± 1.50 | −4.50 ± 0.27 | 0.997 | |
| Matrix | 18.10 ± 0.90 | 7.56 ± 0.42 | 0.9902 | |
| Analyte a | Enrichment Factor b | Recovery c (%) | Intra-Day Precision d (RSD%) | Inter-Day Precision d (RSD%) | Determination Coefficient e (R2) | LOD b (µg L−1) | LOQ b (µg L−1) |
|---|---|---|---|---|---|---|---|
| AZX | 60 | 78 | 8 | 11 | 0.9968 | 0.00070 | 0.0024 |
| BSC | 60 | 78 | 6 | 6 | 0.9977 | 0.0050 | 0.036 |
| BPR | 65 | 86 | 5 | 9 | 0.9955 | 0.0097 | 0.032 |
| CPS | 59 | 77 | 8 | 15 | 0.9992 | 0.16 | 0.54 |
| CPM | 82 | 100 | 8 | 11 | 0.9865 | 1.0 | 1.5 |
| CLF | 63 | 82 | 6 | 10 | 0.9979 | 0.014 | 0.050 |
| DOD | 43 | 56 | 11 | 12 | 0.9805 | 1.0 | 3.0 |
| FLD | 86 | 100 | 14 | 13 | 0.9777 | 1.6 | 5.0 |
| HXT | 62 | 81 | 5 | 9 | 0.9979 | 0.0096 | 0.032 |
| MXF | 68 | 88 | 4 | 5 | 0.9932 | 0.030 | 0.15 |
| MYC | 57 | 74 | 5 | 5 | 0.9987 | 0.016 | 0.050 |
| PEN | 61 | 79 | 3 | 4 | 0.9970 | 0.0083 | 0.028 |
| PYR | 64 | 83 | 4 | 6 | 0.9873 | 0.0054 | 0.018 |
| PRD | 51 | 66 | 4 | 7 | 0.9953 | 0.018 | 0.060 |
| PPF | 58 | 75 | 4 | 7 | 0.9961 | 0.0050 | 0.020 |
| PRO | 62 | 81 | 3 | 3 | 0.9803 | 0.10 | 0.34 |
| STM | 58 | 76 | 6 | 6 | 0.9957 | 0.0097 | 0.032 |
| TEB | 65 | 84 | 3 | 5 | 0.9985 | 0.018 | 0.030 |
| TBF | 68 | 88 | 6 | 8 | 0.9902 | 0.030 | 0.22 |
| Method | Matrix | Common Analytes | Enrichment Factor | Recovery % | Precision (RSD %) | LOD/LOQ#(µgL−1) | Type and Volume of Solvents | Ref. |
|---|---|---|---|---|---|---|---|---|
| DLLME- GC/MS | ultrapure water with 40% ethanol (5 mL) (5 mL) | CPS | between 15 and 20 for all the analytes | 80 | 11 | 0.1/0.34 | Extra: tetrachloroethane (400 µL) Dispb: endogenous ethanol (2000 µL) diluted with water (7.5 mL) | [13] |
| CPM | 109 | 2 | 0.07/0.22 | |||||
| MYC | 100 | 11 | 0.2/0.80 | |||||
| TEB | 68 | 9 | 1.4/4.7 | |||||
| (10 µg L−1 spike level) | ||||||||
| DLLME- GC/MS | white wine (5 mL) | FLD | 66 | 108 | 8.2 | n.d./0.8 | Extra: 1-undecanol (50 µL) Dispb: acetone (500 µL) | [14] |
| PEN | 72 | 100 | 6.8 | n.d./0.3 | ||||
| PRO | 71 | 107 | 7.1 | n.d./1 | ||||
| TEB | 68 | 102 | 6.8 | n.d./0.6 | ||||
| (10 µg L−1 spike level) | ||||||||
| DLLME- GC/MS | white wine (2.5 mL) water diluted to 5 mL | FLD | 1254 | 82 | 4.3 | 0.022/0.074 | Extra: 1-octanol (11 µL) | [15] |
| TEB | 1116 | 74 | 1.3 | 0.010/0.032 | ||||
| (5 µg L−1 spike level) | ||||||||
| QuEChERS-GC/MS | white wine (10 mL) | BPR | n.d. | 92 | 11 | n.d./20 | Acetonitrile (5 mL) + NaCl (3 g), MgSO4 (4 g), PSA (50 mg) | [31] |
| CPS | n.d. | 84 | 12 | n.d./30 | ||||
| CPM | n.d. | 93 | 10 | n.d./20 | ||||
| PRD | n.d. | 87 | 9 | n.d./7 | ||||
| PRO | n.d. | 91 | 5 | n.d./40 | ||||
| TEB | n.d. | 83 | 6 | n.d./40 | ||||
| (50 µg L−1 spike level) | ||||||||
| DLLME- HPLC/MS | white wine (10 mL) | BPR | 65 | 86 | 5 | 0.0097/0.032 | Extra: MEN:BHT (3:1)(150 µL) Dispb: endogenousethanol + addedethanol(total 1850 µL) | This work |
| CPS | 59 | 77 | 8 | 0.16/0.54 | ||||
| CPM | 82 | 100 | 8 | 1.0/1.5 | ||||
| MYC | 57 | 74 | 5 | 0.016/0.050 | ||||
| FLD | 86 | 100 | 14 | 1.6/5.0 | ||||
| PEN | 61 | 79 | 3 | 0.0083/0.028 | ||||
| PRD | 51 | 66 | 4 | 0.018/0.060 | ||||
| PRO | 62 | 81 | 3 | 0.10/0.34 | ||||
| TEB | 65 | 84 | 3 | 0.018/0.030 | ||||
| (5µg L−1 spike level) |
| Analyte | Concentration (µgL−1) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Moscato | Prosecco 1 | Prosecco 2 | Chenin Blanc | Pecorino Bio | Sauvignon Bio | Rosé | Chieti Bio | Montepulciano Bio | Cabernet | Negroamaro | MRL | |
| AZX | 0.17 | 1.1 | 0.16 | 0.090 | n.d. | LOD | <LOD | LOD | LOD | n.d. | 0.15 | 3000 |
| BSC | 1.1 | 4.0 | 10 | 1.2 | <LOD | LOD | 0.59 | LOD | LOD | n.d. | 0.54 | 5000 |
| BPR | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 10 |
| CPS | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 10 |
| CPM | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 10 |
| CLF | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 1000 |
| DOD | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 10 |
| FLD | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 4000 |
| HXT | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 1000 |
| MXF | 2.4 | 3.9 | 1.9 | <LOD | 0.29 | n.d. | 14.4 | 0.30 | 1.5 | <LOD | 9.6 | 1000 |
| MYC | 0.39 | <LOD | LOD | <LOQ | <LOQ | n.d. | n.d. | n.d. | n.d. | 0.15 | 0.36 | 1500 |
| PEN | LOD | n.d. | n.d. | <LOQ | n.d. | n.d. | LOQ | n.d. | n.d. | n.d. | LOQ | 500 |
| PYR | n.d. | n.d. | n.d. | n.d. | n.d. | <LOD | n.d. | n.d. | n.d. | n.d. | n.d. | 2000 |
| PRD | n.d. | n.d. | LOD | LOD | <LOD | n.d. | <LOD | LOD | LOD | <LOQ | LOD | 10 |
| PPF | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 50 |
| PRO | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 300 |
| STM | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 2000 |
| TEB | 0.71 | n.d. | <LOQ | 2.1 | LOD | n.d. | 0.060 | <LOD | <LOD | <LOD | 0.33 | 1000 |
| TBF | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Bosco, C.; Mariani, F.; Gentili, A. Hydrophobic Eutectic Solvent-Based Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Pesticides in Wine. Molecules 2022, 27, 908. https://doi.org/10.3390/molecules27030908
Dal Bosco C, Mariani F, Gentili A. Hydrophobic Eutectic Solvent-Based Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Pesticides in Wine. Molecules. 2022; 27(3):908. https://doi.org/10.3390/molecules27030908
Chicago/Turabian StyleDal Bosco, Chiara, Francesca Mariani, and Alessandra Gentili. 2022. "Hydrophobic Eutectic Solvent-Based Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Pesticides in Wine" Molecules 27, no. 3: 908. https://doi.org/10.3390/molecules27030908
APA StyleDal Bosco, C., Mariani, F., & Gentili, A. (2022). Hydrophobic Eutectic Solvent-Based Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Pesticides in Wine. Molecules, 27(3), 908. https://doi.org/10.3390/molecules27030908