
A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase

 ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
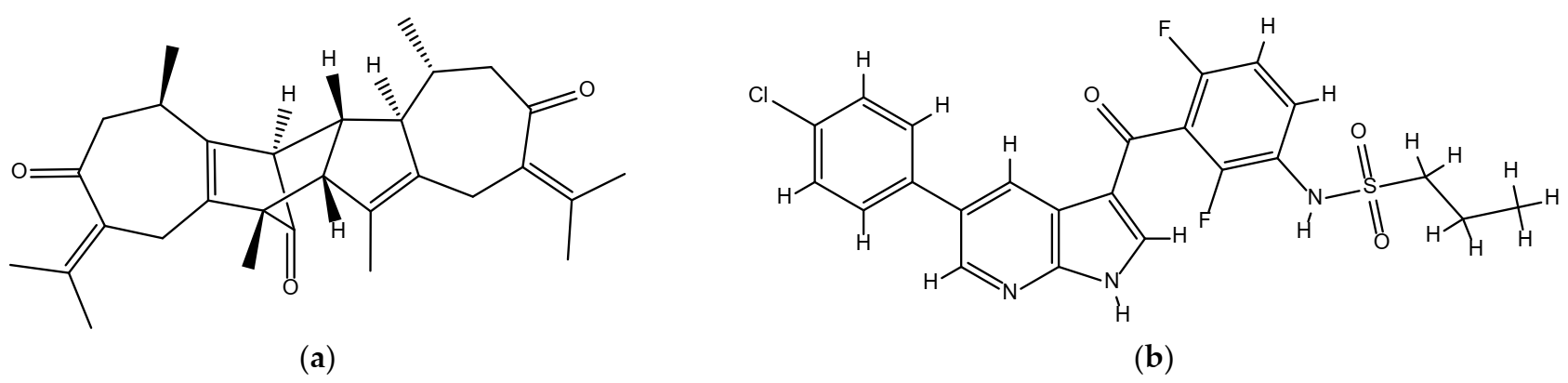
2.1. Chemical Structures of Vieloplain F and Vemurafenib
2.2. Estimation of Activity Spectra for Substances (PASS)
2.3. Toxicological and Pharmacokinetic Assets
2.3.1. Pharmacokinetic Characteristics
2.3.2. Toxicity Assessment
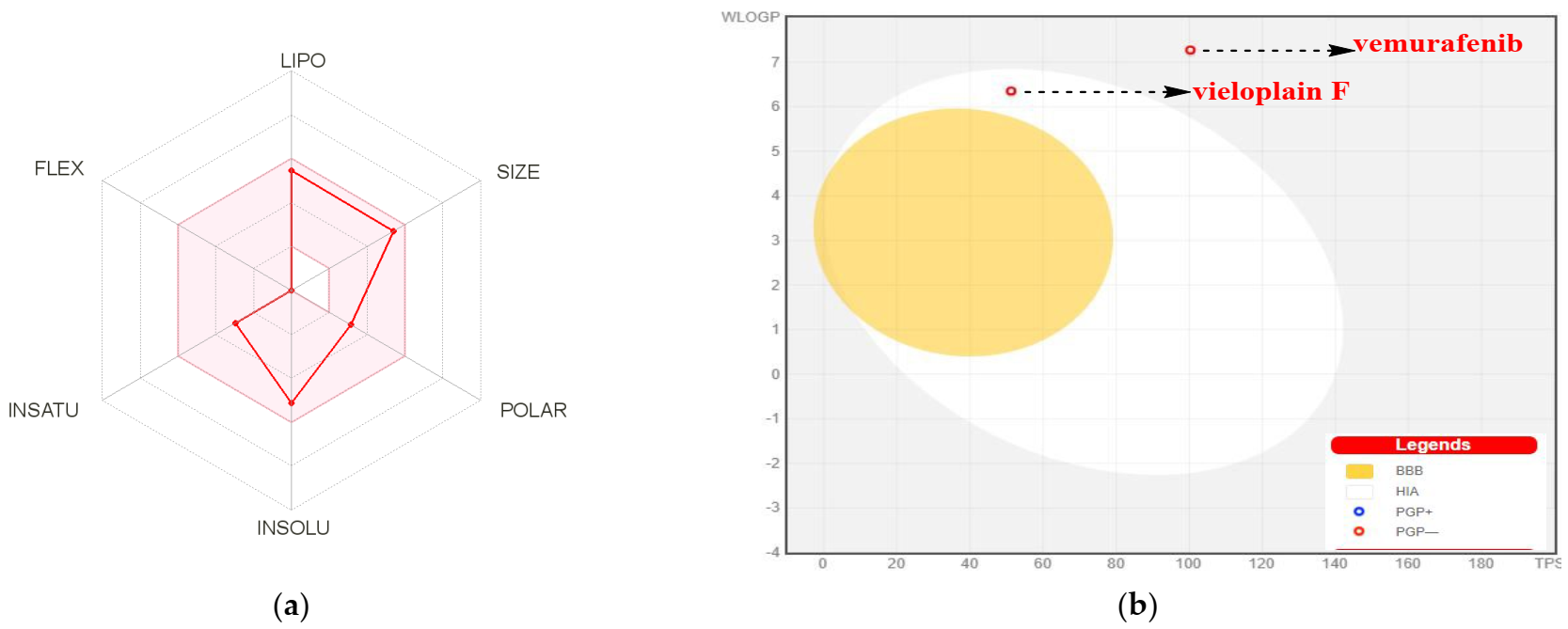
2.4. Drug-Likeness Prediction
2.5. Prediction of the Bioactivity Score
2.6. Prediction of Cardiac Toxicity
2.7. Biomolecular Macromolecules: Epoxidation and Reactivity Prediction
2.8. Prediction of Endocrine Disruption Potential
2.9. Prediction of Cell Line Cytotoxicity
2.10. Docking of the Compounds with B-Raf Kinase Structure (PDB: 3OG7)
2.10.1. Structural Analysis of B-Raf Kinase
2.10.2. Binding Energy Evaluation of the Compounds
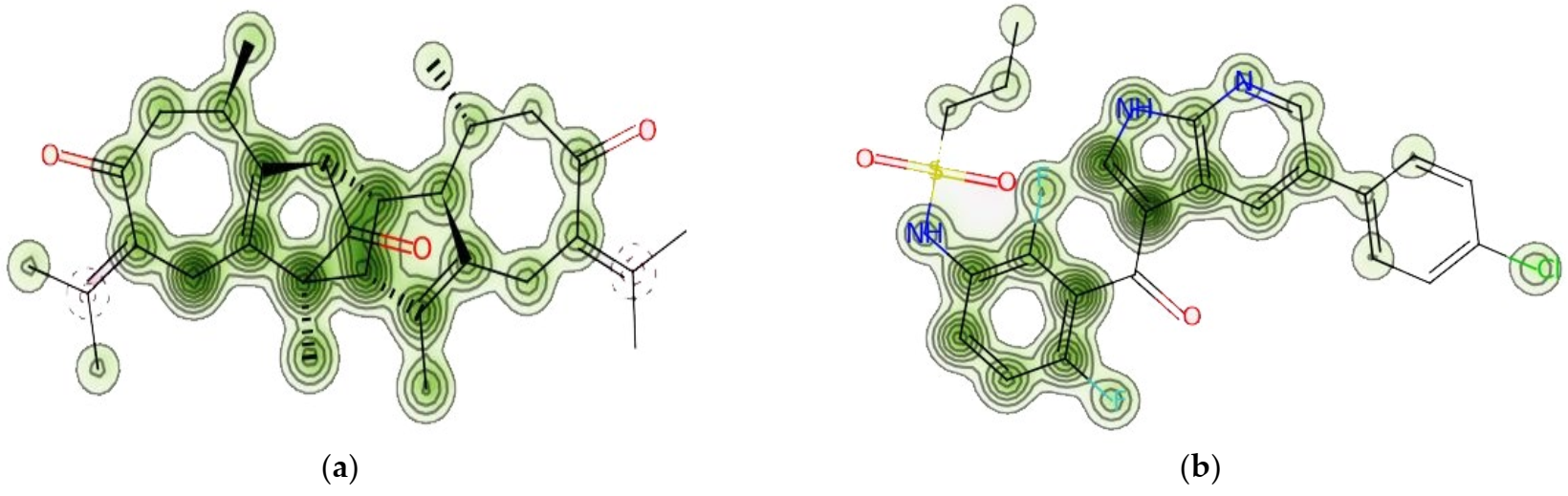
2.10.3. Protein–Ligand Complex Analysis
2.11. Molecular Dynamics and Simulation
2.11.1. RMSD Analysis
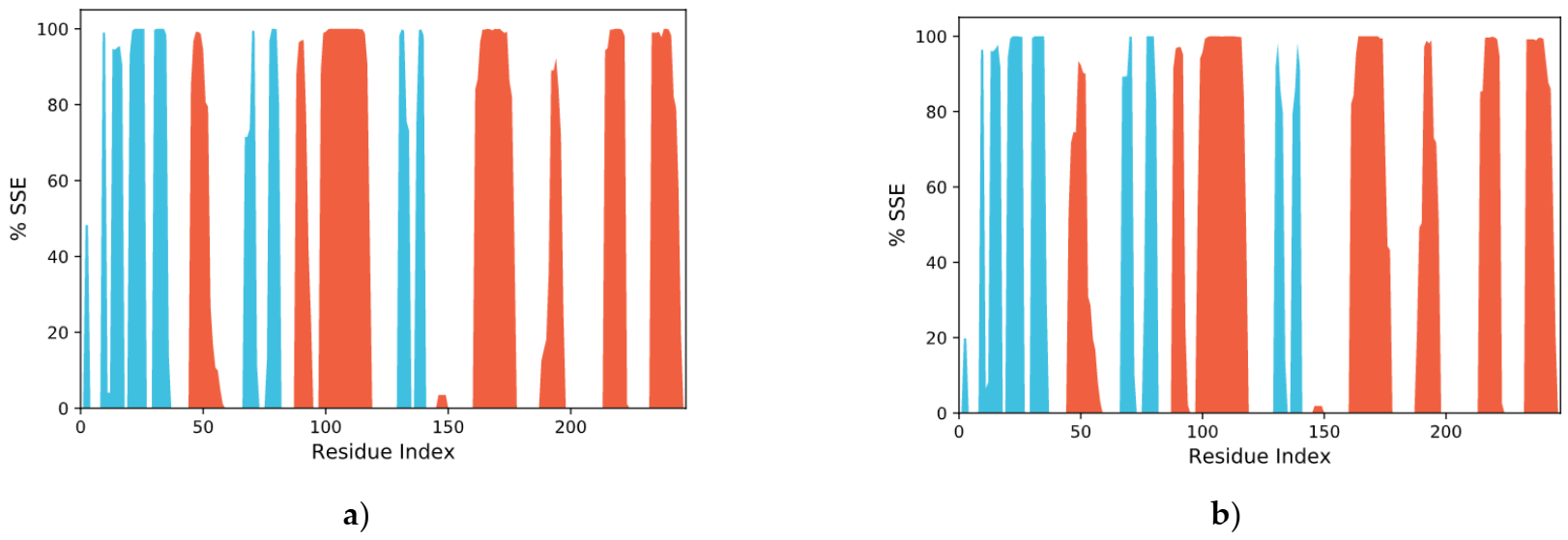
2.11.2. RMSF Assay
2.11.3. Protein–Ligand Interaction
2.11.4. RoG Analysis
2.12. MM-GBSA Calculations
3. Discussion
4. Materials and Methods
4.1. Isolation of Guaiane Dimer Vieloplain F
4.2. Prediction of Activity Spectra for Substances (PASS)
4.3. ADMET Analysis
4.4. Prediction of Acute Rat Toxicity and Environmental Toxicity
4.5. Prediction of Drug-Likeness
4.6. Bioactivity Score Prediction
4.7. Prediction of Cardiac Toxicity
4.8. Prediction of Epoxidation and Reactivity to Biological Macromolecules
4.9. Prediction of Endocrine Disruption Potential
4.10. Prediction of Cell Line Cytotoxicity
4.11. Preparation, Analysis, Retrieval, and Visualization of Protein and Ligand Structures
4.12. Molecular Docking
4.13. Molecular Dynamics Simulations
4.14. MMGBSA Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassan, S.S.u.; Muhammad, I.; Abbas, S.Q.; Hassan, M.; Majid, M.; Jin, H.Z.; Bungau, S. Stress driven discovery of natural products from actinobacteria with anti-oxidant and cytotoxic activities including docking and admet properties. Int. J. Mol. Sci. 2021, 22, 11432. [Google Scholar] [CrossRef] [PubMed]
- Asmat, S.; Shah, A.; Shams, S.; Bungau, S.; Si, Y.; Xu, H.; Rahman, H.; Behl, T.; Gitea, D.; Pavel, F.; et al. Chemically Diverse and Biologically Active Secondary Metabolites from Marine Phylum chlorophyta. Mar. Drugs 2020, 18, 493. [Google Scholar] [CrossRef]
- Khan, I.; Abbas, T.; Anjum, K.; Abbas, S.Q.; Shagufta, B.I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.u. Antimicrobial potential of aqueous extract of Camellia sinensis against representative microbes. Pak. J. Pharm. Sci. 2019, 32, 631–636. [Google Scholar] [PubMed]
- Hassan, S.S.u.; Ishaq, M.; Zhang, W.; Jin, H.-Z. An overview of the mechanisms of marine fungi-derived antiinflammatory and anti-tumor agents and their novel role in drug targeting. Curr. Pharm. Des. 2021, 27, 2605–2614. [Google Scholar] [CrossRef]
- Choi, W.K.; El-Gamal, M.I.; Choi, H.S.; Baek, D.; Oh, C.H. New diarylureas and diarylamides containing 1,3,4-triarylpyrazole scaffold: Synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur. J. Med. Chem. 2011, 46, 5754–5762. [Google Scholar] [CrossRef]
- Naves, L.B.; Almeida, L.; Ramakrishna, S. Understanding the Microenvironment of Melanoma Cells for the Development of Target Drug Delivery Systems. Cit. EMJ Oncol. 2017, 5, 85–92. [Google Scholar]
- Morris, V.; Kopetz, S. BRAF inhibitors in clinical oncology. F1000Prime Rep. 2013, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.S.u.; Jin, H.Z.; Abu-Izneid, T.; Rauf, A.; Ishaq, M.; Suleria, H.A.R. Stress-driven discovery in the natural products: A gateway towards new drugs. Biomed. Pharmacother. 2019, 109, 459–467. [Google Scholar] [CrossRef]
- Xie, Y.G.; Zhao, X.C.; Hassan, S.S.u.; Zhen, X.Y.; Muhammad, I.; Yan, S.K.; Yuan, X.; Li, H.L.; Jin, H. One new sesquiterpene and one new iridoid derivative from Valeriana amurensis. Phytochem. Lett. 2019, 32, 6–9. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhu, S.; Wu, G.; Hassan, S.S.u.; Xie, Y.; Ishaq, M.; Sun, Y.; Yan, S.K.; Qian, X.P.; Jin, H. Chemical Constituents of Vernonia parishii. Chem. Nat. Compd. 2020, 56, 134–136. [Google Scholar] [CrossRef]
- Shams, S.; Zhang, W.; Jin, H.; Basha, S.H.; Priya, S.V.S.S. In-silico anti-inflammatory potential of guaiane dimers from Xylopia vielana targeting COX-2. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Xie, Y.G.; Yan, R.; Zhong, X.; Piao, H.; Muhammad, I.; Ke, X.; Yan, S.; Guo, Y.; Jin, H.Z.; Zhang, W.D. Xylopins A-F, six rare guaiane dimers with three different connecting modes from: Xylopia vielana. RSC Adv. 2019, 9, 9235–9242. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.G.; Guo, Y.G.; Wu, G.J.; Zhu, S.L.; Cheng, T.F.; Zhang, Y.; Yan, S.K.; Jin, H.Z.; Zhang, W.D. Xylopsides A-D, four rare guaiane dimers with two unique bridged pentacyclic skeletons from: Xylopia vielana. Org. Biomol. Chem. 2018, 16, 7030–7034. [Google Scholar] [CrossRef]
- Xie, Y.; Zhong, X.; Xiao, Y.; Zhu, S.; Muhammad, I.; Yan, S.; Jin, H.; Zhang, W. Vieloplains A-G, seven new guaiane-type sesquiterpenoid dimers from Xylopia vielana. Bioorg. Chem. 2019, 88, 102891. [Google Scholar] [CrossRef]
- Hassan, S.S.u.; Shah, S.A.A.; Pan, C.; Fu, L.; Cao, X.; Shi, Y.; Wu, X.; Wang, K.; Wu, B. Production of an antibiotic enterocin from a marine actinobacteria strain H1003 by metal-stress technique with enhanced enrichment using response surface methodology. Pak. J. Pharm. Sci. 2017, 30, 313–324. [Google Scholar]
- Marrero-Ponce, Y.; Siverio-Mota, D.; Gálvez-Llompart, M.; Recio, M.C.; Giner, R.M.; García-Domnech, R.; Torrens, F.; Arán, V.J.; Cordero-Maldonado, M.L.; Esguera, C.V.; et al. Discovery of novel anti-inflammatory drug-like compounds by aligning in silico and in vivo screening: The nitroindazolinone chemotype. Eur. J. Med. Chem. 2011, 46, 5736–5753. [Google Scholar] [CrossRef]
- DiMasi, J.A. Success rates for new drugs entering clinical testing in the United States. Clin. Pharmacol. Ther. 1995, 58, 1–14. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Kolšek, K.; Mavri, J.; Sollner Dolenc, M.; Gobec, S.; Turk, S. Endocrine disruptome—An open source prediction tool for assessing endocrine disruption potential through nuclear receptor binding. J. Chem. Inf. Modeling 2014, 54, 1254–1267. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. In silico Design of Novel HIV-1 NNRTIs Based on Combined Modeling Studies of Dihydrofuro[3,4-d]pyrimidines. Front. Chem. 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Araújo Neto, M.F.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Modeling 2018, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; da Silva, C.H.T.P.; et al. Identification of novel chemical entities for adenosine receptor type 2a using molecular modeling approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 43, 3–26. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 46, 3164–3170. [Google Scholar] [CrossRef]
- Glass, C.K.; Ogawa, S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 44–55. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Druzhilovskiy, D.S.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Dmitriev, A.V.; Pogodin, P.V.; Poroikov, V.V. Computer-aided prediction of biological activity spectra for chemical compounds: Opportunities and limitation. Biomed. Chem. Res. Methods 2018, 1, e00004. [Google Scholar] [CrossRef] [Green Version]
- Spengler, E.K.; Kleiner, D.E.; Fontana, R.J. Vemurafenib-induced granulomatous hepatitis. Hepatology 2017, 65, 745–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, J.; Yan, A.T.; Cramarossa, G.; Chan, K.K.W. Chemotherapy-induced cardiotoxicity: Detection, prevention, and management. Can. J. Cardiol. 2014, 30, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The hERG K+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.; Ostergaard, G.; Larsen, J.C. Toxicological Risk Assessment of Chemicals: A Practical Guide, 1st ed.; CRC Press: Boca Raton, FL, USA, 2008; ISBN 1420006940. [Google Scholar]
- Hughes, T.B.; Miller, G.P.; Swamidass, S.J. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent. Sci. 2015, 1, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.B.; Le Dang, N.; Miller, G.P.; Swamidass, S.J. Modeling reactivity to biological macromolecules with a deep multitask network. ACS Cent. Sci. 2016, 2, 529–537. [Google Scholar] [CrossRef] [PubMed]
- De Coster, S.; van Larebeke, N. Endocrine-Disrupting Chemicals: Associated Disorders and Mechanisms of Action. J. Environ. Public Health 2012, 2012, 713696. [Google Scholar] [CrossRef]
- Braga, R.C.; Alves, V.M.; Silva, M.F.B.; Muratov, E.; Fourches, D.; Lião, L.M.; Tropsha, A.; Andrade, C.H. Pred-hERG: A Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform. 2015, 34, 698–701. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR modelling of rat acute toxicity on the basis of PASS prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Roy, S.; Samant, L.R.; Chowdhary, A. In silico pharmacokinetics analysis and ADMET of phytochemicals of Datura metel Linn. and Cynodon dactylon Linn. J. Chem. Pharm. Res. 2015, 11, 385–388. [Google Scholar]
- Matlock, M.K.; Hughes, T.B.; Swamidass, S.J. XenoSite server: A web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: Review on chemdraw, chemwindow, ISIS/draw, and chemsketch. J. Chem. Inf. Comput. Sci. 2004, 44, 1886–1890. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, Y.; Yu, X.-F.; Tan, Q.-Q.; Liang, S.-S.; Li, T.; Zhang, H.; Shaw, P.-C.; Wang, J.; Hu, C. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: Molecular dynamics simulation and MM-GBSA calculation. Comput. Biol. Chem. 2020, 85, 107241. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Activities | Pa | Pi |
|---|---|---|
| Anti-neoplastic | 0.862 | 0.006 |
| Anti-leukemic | 0.592 | 0.009 |
| Testosterone 17beta-dehydrogenase (NADP+) inhibitor | 0.589 | 0.094 |
| Properties | Parameters | Vieloplain F | Vemurafenib |
|---|---|---|---|
| Physicochemical Properties | MW a (g/mol) | 446.62 | 489.92 |
| Rotatable bonds | 0 | 7 | |
| HBA b | 3 | 6 | |
| HBD c | 0 | 2 | |
| Fraction Csp3 | 0.63 | 0.13 | |
| TPSA d | 51.21 | 100.30 | |
| Lipophilicity Log Po/w | iLOGP | 3.93 | 3.04 |
| XLOGP3 | 4.02 | 4.97 | |
| MLOGP | 6.35 | 3.41 | |
| Consensus | 5.07 | 4.89 |
| Properties | Parameters | Vieloplain F | Vemurafenib |
|---|---|---|---|
| Absorption | Water solubility | −5.971 | −4.656 |
| GI a | 100 | 98.45 | |
| Log Kp (skin permeation) cm/s | −6.17 | −5.76 | |
| Distribution | BBB b | −0.37 | −1.686 |
| CNS permeation (Log PS) | −1.027 | −2.976 | |
| VD c (human) | −0.013 | −0.461 | |
| Metabolism CYP2D6 | CYP1A2 inhibitor | No | No |
| CYP2C9 inhibitor | No | Yes | |
| CYP2C19 inhibitor | No | Yes | |
| CYP3A4 inhibitor | No | Yes | |
| CYP2D6 inhibitor | No | No | |
| Excretion | Total Clearance (log mL/min/kg) | 0.053 | 0.136 |
| Renal OCT2 substrate | No | No |
| Parameters | Vieloplain F | Vemurafenib |
|---|---|---|
| Ames Toxicity | No | No |
| Max. Tolerated Dose (human) (log mg/kg/day) | 0.013 | 0.601 |
| hERG I Inhibitor | No | No |
| hERG II Inhibitor | Yes | Yes |
| Oral Toxicity (LD50) (mg/kg) | 1640 | 2316 |
| Oral Toxicity classification * | IV | V |
| Hepatotoxicity | No | Yes |
| Skin Sensitization | No | No |
| Bioaccumulation Factor Log10 (BCF) | 2.489 | 0.674 |
| Daphnia magna LC50 − Log10 (mol/L) | 7.127 | 6.969 |
| Fathead Minnow LC50·Log10 (mmol/L) | −4.972 | −4.154 |
| Tetrahymena pyriformis IGC50 − Log10 (mol/L) | 1.998 | 2.203 |
| Compounds | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|
| Vemurafenib | 0.45 | 0.25 | 0.64 | 0.02 | 0.12 | 0.34 |
| Vieloplain F | −0.12 | −0.18 | −0.54 | 0.17 | −0.04 | 0.06 |
| Compound | Cell Line | Cell Line Full Name | Tissue | Tumor Type | Pa | Pi |
|---|---|---|---|---|---|---|
| Vemurafenib | SK-MEL-28 | Melanoma | Skin | Melanoma | 0.618 | 0.010 |
| A2058 | Melanoma | Skin | Melanoma | 0.581 | 0.004 | |
| M14 | Melanoma | Skin | Melanoma | 0.546 | 0.012 | |
| PA-1 | Ovarian carcinoma | Ovarium | Carcinoma | 0.520 | 0.005 | |
| Vieloplain F | SK-MEL-2 | Melanoma | Skin | Melanoma | 0.722 | 0.006 |
| K562 | Erythroleukemia | Hematopoietic and lymphoid tissue | Leukemia | 0.598 | 0.015 |
| Compound Name | Chemical Structure | Binding Energy (kcal/mol) | Amino Acid Residues Involved in the Bonding |
|---|---|---|---|
| Vieloplain F | 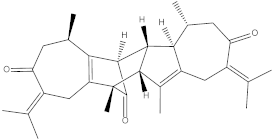 | −11.8 | Ser164, Asp165, Asp125, Lys127, Trp150, Ser147, Ile148, Met151, Val155. |
| Vemurafenib |  | −10.2 | Asp143, Lys127, Asn129, Ser88, Phe132, Ser87, Cys84, Trp83, Ala33, Ile15, Ile79, Lys35, Phe20. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.S.u.; Abbas, S.Q.; Ali, F.; Ishaq, M.; Bano, I.; Hassan, M.; Jin, H.-Z.; Bungau, S.G. A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase. Molecules 2022, 27, 917. https://doi.org/10.3390/molecules27030917
Hassan SSu, Abbas SQ, Ali F, Ishaq M, Bano I, Hassan M, Jin H-Z, Bungau SG. A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase. Molecules. 2022; 27(3):917. https://doi.org/10.3390/molecules27030917
Chicago/Turabian StyleHassan, Syed Shams ul, Syed Qamar Abbas, Fawad Ali, Muhammad Ishaq, Iqra Bano, Mubashir Hassan, Hui-Zi Jin, and Simona G. Bungau. 2022. "A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase" Molecules 27, no. 3: 917. https://doi.org/10.3390/molecules27030917
APA StyleHassan, S. S. u., Abbas, S. Q., Ali, F., Ishaq, M., Bano, I., Hassan, M., Jin, H.-Z., & Bungau, S. G. (2022). A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase. Molecules, 27(3), 917. https://doi.org/10.3390/molecules27030917