
Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study


 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results
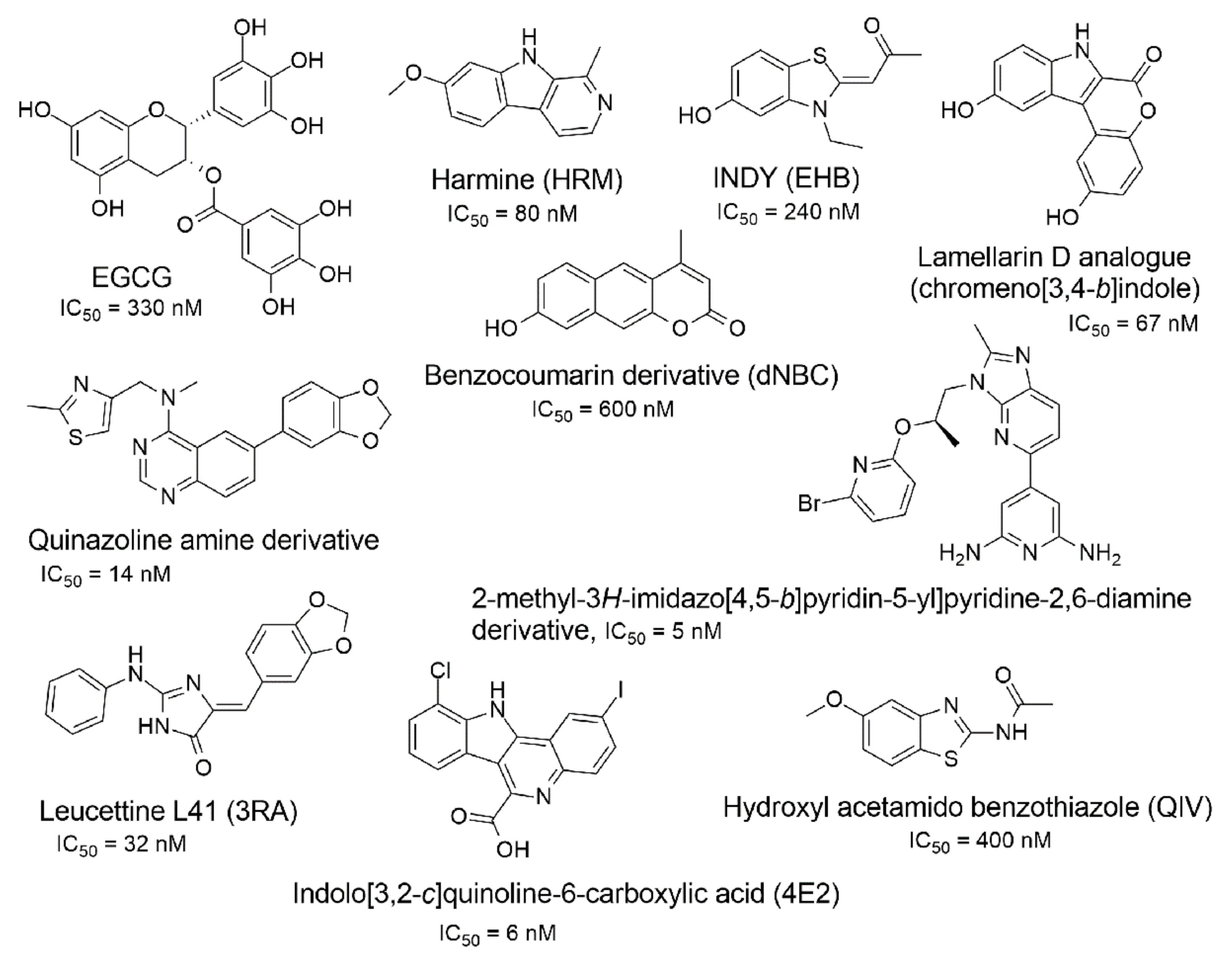
2.1. Docking Library
2.2. Human DYRK1A and Related Protein Kinases
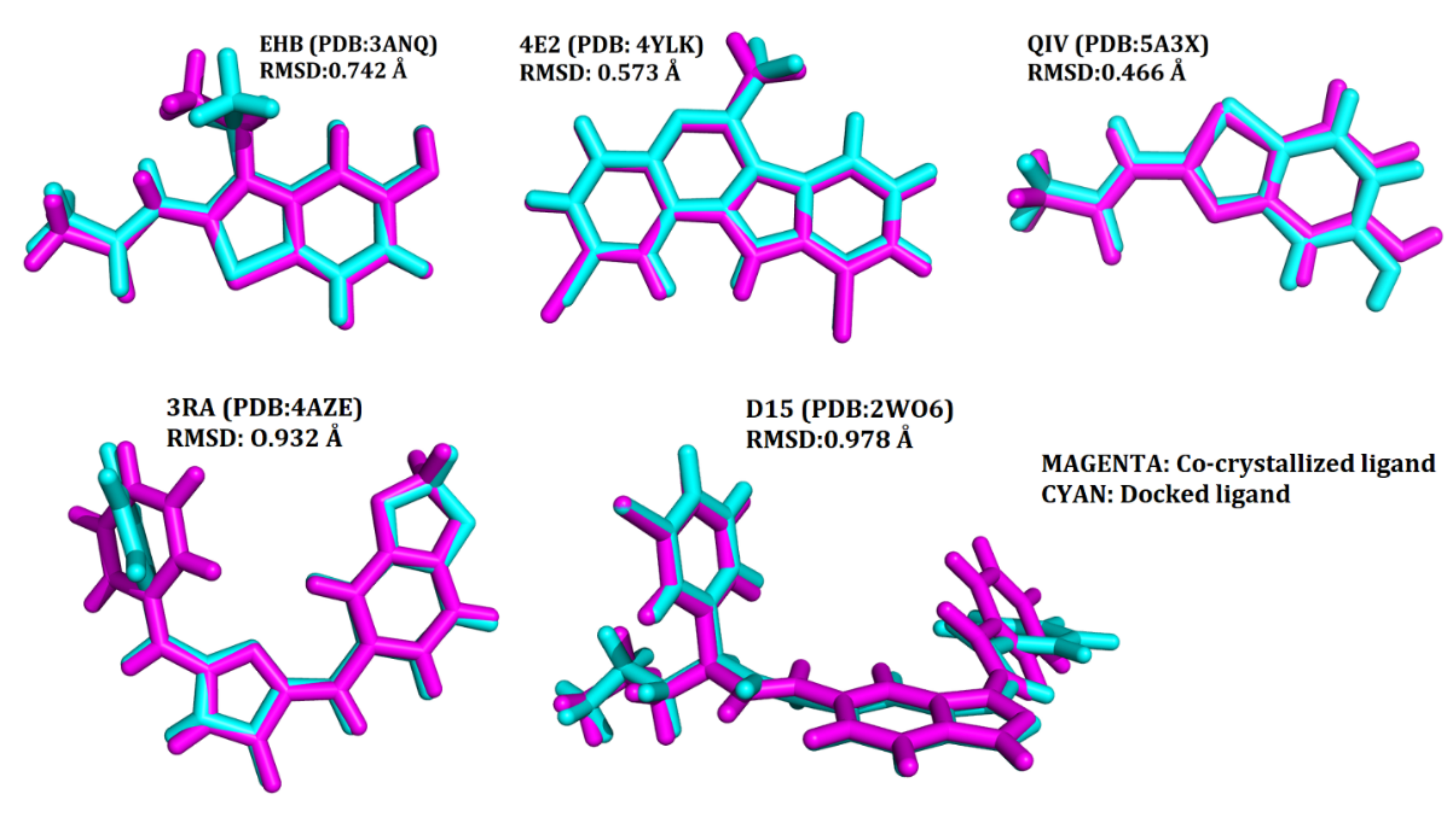
2.3. Validation of the Docking Protocol
2.4. Docking-Based Virtual Screening
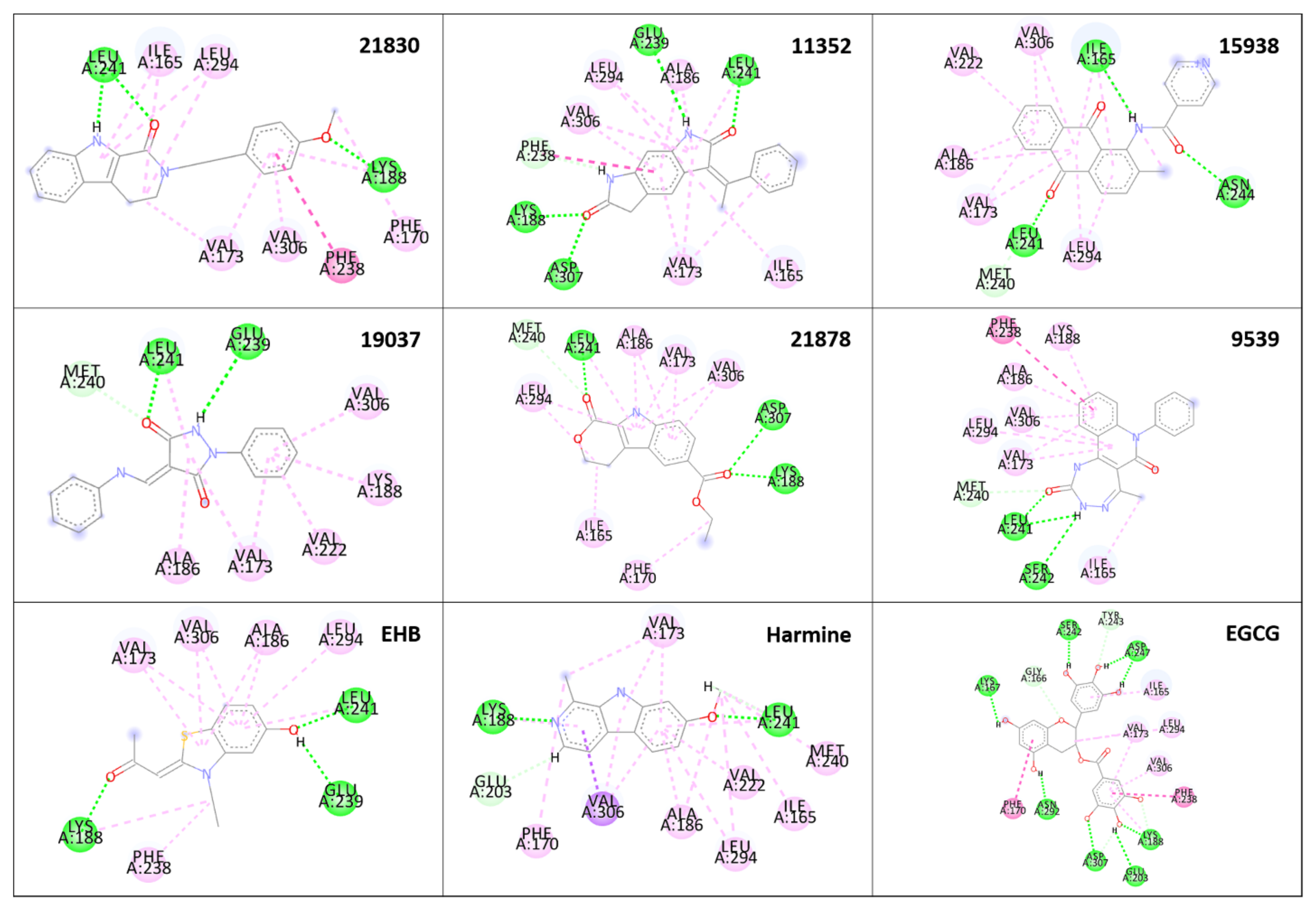
2.5. Analysis of the Binding Pattern
2.6. MM/GBSA Binding Free Energy Calculations
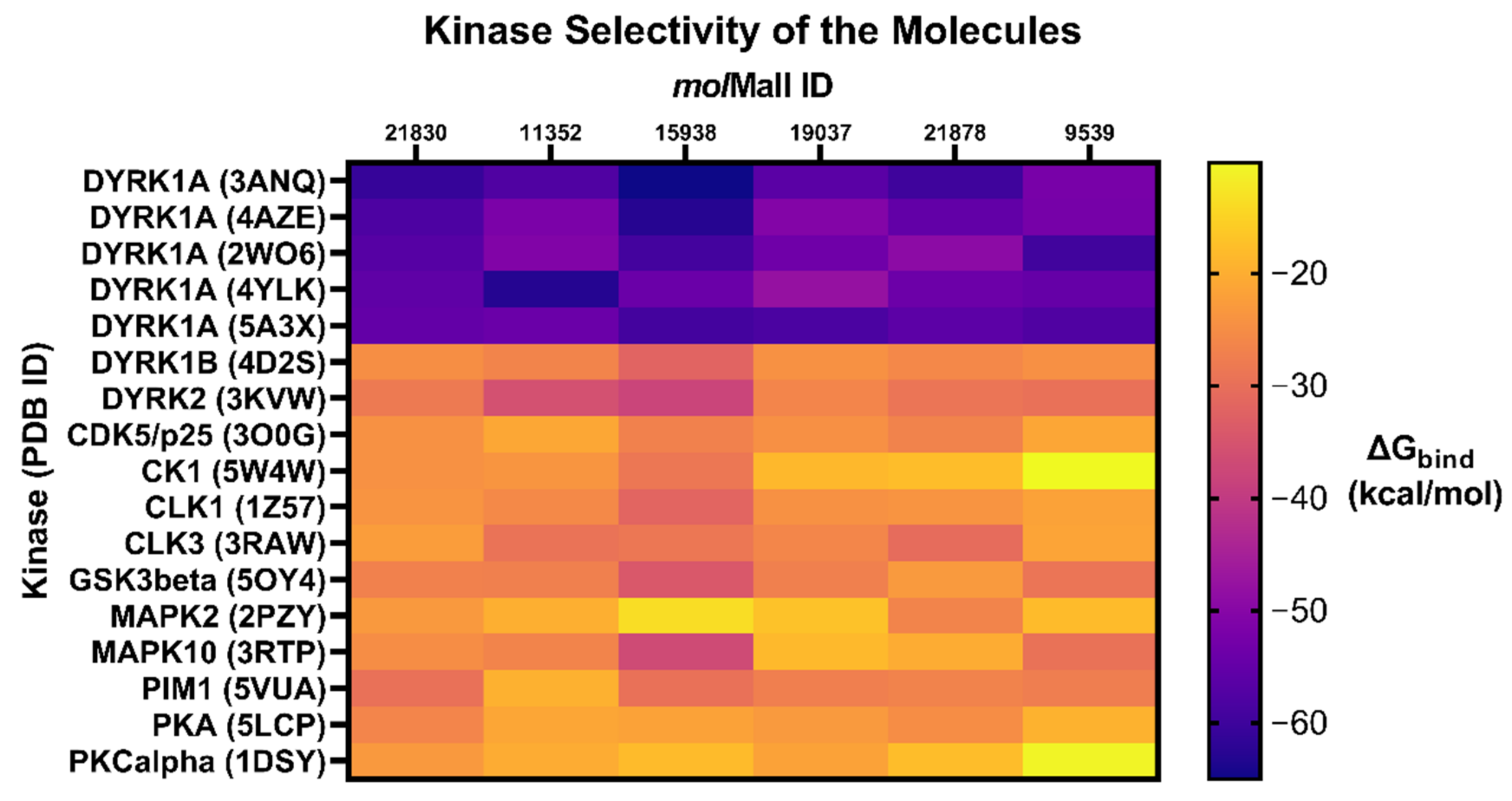
2.7. Glide XP Docking with Related Protein Kinases
2.8. Physicochemical and Pharmacokinetic Properties
2.9. Toxicity Studies
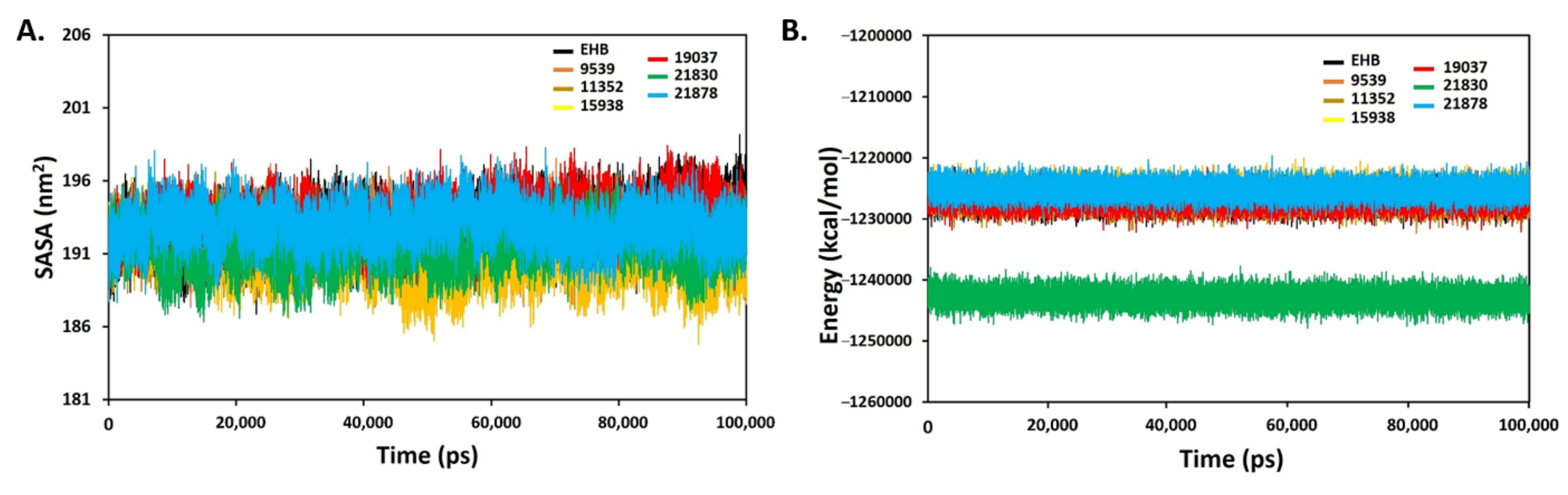
2.10. Molecular Dynamics (MD) Simulation
2.11. In Silico Bioactivity Prediction
2.12. Structure Similarity Comparison
2.13. Synthetic Accessibility (SA) Prediction
3. Discussion
4. Materials and Methods
4.1. Preparation of the Docking Library
4.2. DYRK1A and Related Protein Kinases: Selection, Preparation and Grid Generation
4.3. Validation of the Docking Protocol
4.4. Docking-Based Virtual Screening
4.5. Analysis of the Binding Pattern
4.6. MM/GBSA Binding Free Energy Calculations
4.7. Glide XP Docking with Related Protein Kinases
4.8. Physicochemical and Pharmacokinetic Properties
4.9. Toxicity Studies
4.10. Molecular Dynamics (MD) Simulation
4.11. In Silico Bioactivity Prediction
4.12. Structure Similarity Comparison
4.13. Synthetic Accessibility (SA) Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Tredici, K.D. Where, when, and in what form does sporadic Alzheimer disease begin? Curr. Opin. Neurol. 2012, 25, 708–714. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dementia fact sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 22 September 2020).
- Fink, H.A.; Linskens, E.J.; MacDonald, R.; Silverman, P.C.; McCarten, J.R.; Talley, K.M.C.; Forte, M.L.; Desai, P.J.; Nelson, V.A.; Miller, M.A.; et al. Benefits and Harms of Prescription Drugs and Supplements for Treatment of Clinical Alzheimer-Type Dementia. Ann. Intern. Med. 2020, 172, 656–668. [Google Scholar] [CrossRef] [PubMed]
- US FDA Newsroom. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 7 June 2021).
- Hardy, J. Genetic dissection of neurodegenerative disease. Clin. Neurosci. Res. 2001, 1, 134–141. [Google Scholar] [CrossRef]
- Tirabhschi, P.; Hansen, L.A.; Thai, L.J.; Corey-Bloom, J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 2004, 62, 1984–1989. [Google Scholar] [CrossRef]
- Guedi, F.; Pereira, P.L.; Najas, S.; Barallobre, M.J.; Chabert, C.; Souchet, B.; Sebrie, C.; Verney, C.; Herault, Y.; Arbones, M.; et al. DYRK1A: A master regulatory protein controlling brain growth. Neurobiol. Dis. 2012, 46, 190–203. [Google Scholar]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef]
- Becker, R.E.; Greig, N.H.; Giacobini, E.; Schneider, L.S.; Ferrucci, L. A new roadmap for drug development for Alzheimer’s disease. Nat. Rev. Drug Discov. 2014, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Drachman, D.A. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 372–380. [Google Scholar] [CrossRef]
- Tejedor, F.J.; Hammerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2010, 278, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Aranda, S.; Laguna, A.; de la Luna, S. DYRK family of protein kinases: Evolutionary relationships, biochemical properties, and functional roles. FASEB J. 2011, 25, 449–462. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Yoo, L.; Jung, M.S.; Song, W.J.; Lee, S.H.; Seo, H.; Chung, K.C. Dyrk1A Phosphorylates p53 and Inhibits Proliferation of Embryonic Neuronal Cells. J. Biol. Chem. 2010, 285, 31895–31906. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Wegiel, J.; Kaczmarski, W.; Barua, M.; Kuchna, I.; Nowicki, K.; Wang, K.C.; Wegiel, J.; Yang, S.M.; Frackowiak, J.; Mazur-Kolecka, B.; et al. Link Between DYRK1A Overexpression and Several-Fold Enhancement of Neurofibrillary Degeneration with 3-Repeat Tau Protein in Down Syndrome. J. Neuropathol. Exp. Neurol. 2011, 70, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased Dosage of Dyrk1A Alters Alternative Splicing Factor (ASF)-regulated Alternative Splicing of Tau in Down Syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef] [Green Version]
- Azorsa, D.O.; Robeson, R.H.; Frost, D.; Meechoovet, B.; Brautigam, G.R.; Dickey, C.; Beaudry, C.; Basu, G.D.; Holz, D.R.; Hernandez, J.A.; et al. High-content siRNA screening of the kinome identifies kinases involved in Alzheimer’s disease-related tau hyperphosphorylation. BMC Genom. 2010, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Frost, D.; Meechoovet, B.; Wang, T.; Gately, S.; Giorgetti, M.; Shcherbakova, I.; Dunckley, T. β-Carboline Compounds, Including Harmine, Inhibit DYRK1A and Tau Phosphorylation at Multiple Alzheimer’s Disease-Related Sites. PLoS ONE 2011, 6, e19264. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.S.; Park, J.H.; Ryu, Y.S.; Choi, S.H.; Yoon, S.H.; Kwen, M.Y.; Oh, J.Y.; Song, W.J.; Chung, S.H. Regulation of RCAN1 Protein Activity by Dyrk1A Protein-mediated Phosphorylation. J. Biol. Chem. 2011, 286, 40401–40412. [Google Scholar] [CrossRef] [Green Version]
- Song, W.J.; Song, E.A.C.; Choi, S.H.; Baik, H.H.; Jin, B.K.; Kim, J.H.; Chung, S.H. Dyrk1A-mediated phosphorylation of RCAN1 promotes the formation of insoluble RCAN1 aggregates. Neurosci. Lett. 2013, 554, 135–140. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, S.R.; Cho, H.J.; Lee, H.W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.S.; Kim, M.J.; Son, M.Y.; Seo, H.; Chung, S.H.; et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.S.; Yoon, S.H.; Kwen, M.Y.; Lee, S.Y.; Choi, S.H.; Radnaabazar, C.; Kim, M.K.; Kim, H.; et al. Dyrk1A-mediated phosphorylation of Presenilin 1: A functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sung, J.Y.; Lee, H.J.; Rhim, H.; Hasegawa, M.; Iwatsubo, T.; Min, D.S.; Kim, J.; Paik, S.R.; Chung, K.C. DYRK1A Phosphorylates α-Synuclein and Enhances Intracellular Inclusion Formation. J. Biol. Chem. 2006, 281, 33250–33257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Kyng, A.K.; Kim, N.D.; Chon, Y.S.; Jung, M.S.; Lee, B.J.; Kim, J.H.; Song, W.J. QSAR analysis of pyrazolidine-3,5-diones derivatives as Dyrk1A inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2324–2328. [Google Scholar]
- Akue-Gedu, R.; Debiton, E.; Ferandin, Y.; Meijer, L.; Prudhomme, M.; Anizon, F.; Moreau, P. Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg. Med. Chem. 2009, 17, 4420–4424. [Google Scholar] [CrossRef] [Green Version]
- Chioua, M.; Samadi, A.; Soriano, E.; Lozach, O.; Meijer, L.; Marco-Contelles, J. Synthesis and biological evaluation of 3,6-diamino-1H-pyrazolo[3,4-b]pyridine derivatives as protein kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4566–4569. [Google Scholar] [CrossRef]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef] [Green Version]
- Guedi, F.; Sébrié, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green Tea Polyphenols Rescue of Brain Defects Induced by Overexpression of DYRK1A. PLoS ONE 2009, 4, e4606. [Google Scholar]
- Gockler, N.; Guillermo, J.; Papadopoulos, C.; Soppa, U.; Tejador, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [Green Version]
- Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Synthesis and evaluation of β-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg. Med. Chem. 2011, 19, 134–144. [Google Scholar] [CrossRef]
- Rosenthal, A.S.; Tanega, C.; Shen, M.; Mott, B.T.; Bougie, J.M.; Nguyen, D.T.; Misteli, T.; Auld, D.S.; Maloney, D.J.; Thomas, C.J. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg. Med. Chem. Lett. 2011, 21, 3152–3158. [Google Scholar] [CrossRef] [Green Version]
- Sarno, S.; Mazzorana, M.; Traynor, R.; Ruzzene, M.; Cozza, G.; Pagano, M.A.; Meggio, F.; Zagotto, G.; Battistutta, R.; Pinna, L.A. Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: Role of a nitro group that discriminates between CK2 and DYRK1A. Cell. Mol. Life Sci. 2012, 69, 449–460. [Google Scholar] [CrossRef]
- Neagoi, C.; Vedrenne, E.; Buron, F.; Mérour, J.Y.; Rosca, S.; Bourg, S.; Lozach, O.; Meijer, L.; Baldeyrou, B.; Lansiaux, A.; et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: A novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012, 49, 379–396. [Google Scholar] [CrossRef]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, Cocrystal Structures, and Neuroprotective Properties of Leucettines, a Family of Protein Kinase Inhibitors Derived from the Marine Sponge Alkaloid Leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Soundararajan, M.; Roos, A.K.; Savitsky, P.; Filippakopoulos, P.; Kettenbach, A.N.; Olsen, J.V.; Gerber, S.A.; Eswaran, J.; Knapp, S.; Elkins, J.M. Structures of Down Syndrome Kinases, DYRKs, Reveal Mechanisms of Kinase Activation and Substrate Recognition. Structure 2013, 21, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Falke, H.; Chaikuad, A.; Becker, A.; Loaec, N.; Lozach, O.; Abu Jhaisha, S.; Becker, W.; Jones, P.G.; Preu, L.; Baumann, K.; et al. 10-Iodo-11H-indolo[3,2-c]quinoline-6-carboxylic Acids Are Selective Inhibitors of DYRK1A. J. Med. Chem. 2015, 58, 3131–3143. [Google Scholar] [CrossRef]
- Rothweiler, U.; Stensen, W.; Brandsdal, B.O.; Isaksson, J.; Leeson, F.A.; Eugh, R.A.; Svendsen, J.S.M. Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules. J. Med. Chem. 2016, 59, 9814–9824. [Google Scholar] [CrossRef]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H.; et al. Novel Scaffolds for Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK1A) Inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Sipos, M.; Paczal, A.; Balint, B.; Kun, V.; Foloppe, N.; Dokurno, P.; Massey, A.J.; Walmsley, D.L.; Hubbard, R.E.; et al. Structure-Guided Discovery of Potent and Selective DYRK1A Inhibitors. J. Med. Chem. 2021, 64, 6745–6764. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer–An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Beauchard, A.; Laborie, H.; Rouillard, H.; Lozach, O.; Ferandin, Y.; Guével, R.L.; Gugen-Guillouzo, C.; Meijer, L.; Besson, T.; Thiéry, V. Synthesis and Kinase Inhibitory Activity of Novel Substituted Indigoids. Bioorg. Med. Chem. 2009, 17, 6257–6263. [Google Scholar] [CrossRef]
- Kettle, J.G.; Ballard, P.; Bardelle, C.; Cockerill, M.; Colclough, N.; Critchlow, S.E.; Debreczeni, J.E.; Fairley, G.; Fillery, S.; Graham, M.A.; et al. Discovery and Optimization of a Novel Series of Dyrk1B Kinase Inhibitors to Explore a MEK Resistance Hypothesis. J. Med. Chem. 2015, 58, 2834–2844. [Google Scholar] [CrossRef]
- Ahn, J.S.; Radhakrishnan, M.L.; Mapelli, M.; Choi, S.; Tidor, B.; Cuny, G.D.; Musacchio, A.; Yeh, L.; Kosik, S.K. Defining Cdk5 Ligand Chemical Space with Small Molecule Inhibitors of Tau Phosphorylation. Chem. Biol. 2005, 12, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Wager, T.T.; Galatsis, P.; Chandrasekaran, R.Y.; Butler, T.W.; Li, J.; Zhang, L.; Mente, S.; Subramanyam, C.; Liu, S.; Doran, A.C.; et al. Identification and Profiling of a Selective and Brain Penetrant Radioligand for in Vivo Target Occupancy Measurement of Casein Kinase 1 (CK1) Inhibitors. ACS Chem. Neurosci. 2017, 8, 1995–2004. [Google Scholar] [CrossRef]
- Bullock, A.N.; Das, S.; Debreczeni, J.E.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Henley, Z.A.; Bax, B.D.; Inglesby, L.M.; Champigny, A.; Gaines, S.; Faulder, P.; Le, J.; Thomas, D.A.; Washio, Y.; Baldwin, I.R. From PIM1 to PI3Kδ via GSK3β: Target Hopping through the Kinome. ACS Med. Chem. Lett. 2017, 8, 1093–1098. [Google Scholar] [CrossRef]
- Wu, J.P.; Wang, J.; Abeywardane, A.; Andersen, D.; Emmanuel, M.; Gautschi, E.; Goldberg, D.R.; Kashem, M.A.; Lukas, S.; Mao, W.; et al. The discovery of carboline analogs as potent MAPKAP-K2 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4664–4669. [Google Scholar] [CrossRef] [PubMed]
- Bowers, S.; Truong, A.P.; Neitz, R.J.; Hom, R.K.; Sealy, J.M.; Probst, G.D.; Quincy, D.; Peterson, B.; Chan, W.; Galemmo, R.A.; et al. Design and synthesis of brain penetrant selective JNK inhibitors with improved pharmacokinetic properties for the prevention of neurodegeneration. Bioorg. Med. Chem. Lett. 2011, 21, 5521–5527. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.; Watanabe, H.; Fukuzawa, K.; Parker, L.J.; Okiyama, Y.; Yuki, H.; Yokoyama, S.; Nakano, H.; Tanaka, S.; Honma, T. Theoretical Analysis of Activity Cliffs among Benzofuranone-Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson–Boltzmann Surface Area (FMO+MM-PBSA) Approach. J. Chem. Inf. Model. 2017, 57, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Verdaguer, N.; Corbalan-Garcia, S.; Ochoa, W.F.; Fita, I.; Gomez-Fernandez, J.C. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. EMBO J. 1999, 18, 6329–6338. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Kamaraj, B.; Bogaerts, A. Structure and Function of p53-DNA Complexes with Inactivation and Rescue Mutations: A Molecular Dynamics Simulation Study. PLoS ONE 2015, 10, e0134638. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathoree, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2020, 39, 3760–3770. [Google Scholar] [CrossRef]
- Osborne, R. Myriad stumbles, Wyeth closes on Alzheimer’s. Nat. Biotech. 2008, 26, 841–843. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Extance, A. Alzheimer’s failure raises questions about disease-modifying strategies. Nat. Rev. Drug Discov. 2010, 9, 749–751. [Google Scholar] [CrossRef]
- Haas, C. Strategies, Development, and Pitfalls of Therapeutic Options for Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 28, 241–281. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Zhou, T.; Lafleur, K.; Nevado, C.; Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: A network analysis. Bioinformatics 2010, 26, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Maestro, Version 10.2; Schrödinger LLC.: New York, NY, USA, 2015.
- Glide, Version 6.7; Schrödinger LLC.: New York, NY, USA, 2015.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA Dassault Systèmes. Biovia Discovery Studio Visualizer, Version 20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2020.
- PyMOL Molecular Graphics System, Version 1.8.4.0; Schrödinger LLC.: New York, NY, USA, 2016.
- Prime, Version 4.0; Schrödinger LLC.: New York, NY, USA, 2015.
- QikProp, Version 4.4; Schrödinger LLC.: New York, NY, USA, 2015.
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.molinspiration.com/cgi-bin/properties (accessed on 23 January 2022).
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- Available online: http://ambit.sourceforge.net/reactor.html (accessed on 5 February 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDPI (molMall) ID/Known Inhibitors | Chemical Structures | DYRK1A Crystal Structures (PDB IDs) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3ANQ | 4AZE | 2WO6 | 4YLK | 5A3X | |||||||
| Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | ||
| 21830 | 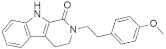 | −11.64 | −61.15 | −10.07 | −58.04 | −11.23 | −56.94 | −11.62 | −55.68 | −11.88 | −55.17 |
| 11352 |  | −10.96 | −57.79 | −11.24 | −51.85 | −10.80 | −50.94 | −10.74 | −63.00 | −10.45 | −54.08 |
| 15938 |  | −10.41 | −65.02 | −10.31 | −62.92 | −8.47 | −59.30 | −8.13 | −54.20 | −8.41 | −59.42 |
| 19037 |  | −9.97 | −56.42 | −8.57 | −50.66 | −8.51 | −53.44 | −9.09 | −47.94 | −10.53 | −58.43 |
| 21878 | 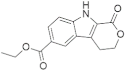 | −9.42 | −59.96 | −10.34 | −55.25 | −8.34 | −49.26 | −9.91 | −53.93 | −8.80 | −56.15 |
| 9539 | 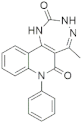 | −8.00 | −52.33 | −10.79 | −52.45 | −9.08 | −59.66 | −10.19 | −54.87 | −10.35 | −57.73 |
| EHB |  | −7.72 | −56.42 | −7.33 | −51.46 | −7.49 | −54.30 | −6.45 | −51.68 | −7.83 | −55.40 |
| Harmine | 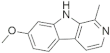 | −7.22 | −49.24 | −6.54 | −54.59 | −7.42 | −46.55 | −7.29 | −49.11 | −8.15 | −48.79 |
| EGCG |  | −10.76 | −64.44 | −12.21 | −70.48 | −13.17 | −63.32 | −11.99 | −65.67 | −11.73 | −65.01 |
| MDPI (molMall) ID/Known Inhibitors | Chemical Structures | DYRK1A (PDB ID: 3ANQ): Polar and Non-Polar Interactions | ||
|---|---|---|---|---|
| H-Bond | Pi-Pi | Alkyl and Pi-Alkyl | ||
| 21830 |  | Lys188—OCH3 (2.28 Å), Leu241—O=C (2.08 Å), Leu241—NH (2.09) | Phe238 | Ile165, Phe170, Val173, Lys188, Leu294, Val306 |
| 11352 |  | Lys188—O=C (2.14 Å), Glu239—HN (2.52 Å), Leu241—O=C (2.03 Å), Asp307—O=C (2.61 Å) | Phe238 | Ile165, Val173, Ala186, Leu294, Val306 |
| 15938 |  | Ile165—HN (2.26 Å), Leu241—O=C (1.99 Å), Asn244—O=C (2.05 Å) | --- | Ile165, Val173, Ala186, Val222, Leu294, Val306 |
| 19037 | 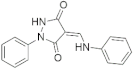 | Glu239—HN (2.84 Å), Leu241—O=C (1.91 Å) | --- | Val173, Ala186, Lys188, Val222, Leu241, Val306 |
| 21878 |  | Lys188—O=C (2.13 Å), Leu241—O=C (1.87 Å), Asp307—O=C (2.89 Å) | --- | Ile165, Phe170, Val173, Ala186, Leu241, Leu294, Val306 |
| 9539 |  | Leu241—O=C (1.86 Å), Leu241—HN (2.44), Ser242—HN (2.98) | Phe238 | Ile165, Val173, Ala186, Lys188, Leu294, Val306 |
| EHB |  | Lys188—O=C (2.72 Å), Glu239—HO (2.15 Å), Leu241—OH (2.00 Å) | --- | Val173, Ala186, Lys188, Phe238, Leu241, Leu294, Val306 |
| Harmine | 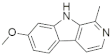 | Lys188—N (1.92 Å), Leu241—OCH3 (2.13 Å) | --- | Ile165, Phe170, Val173, Ala186, Lys188, Val222, Met240, Leu241, Leu294, Val306 |
| EGCG |  | Lys167—HO (1.85 Å), Lys188—OH (1.69 Å), Glu203—HO (2.66 Å), Ser242—HO (1.67 Å), Asp247—HO (1.75 Å), Asp247—HO (1.46 Å), Asn292—HO (1.78 Å), Asp307—OH (2.56 Å) | Phe170, Phe238 | Ile165, Val173, Lys188, Leu294, Val306 |
| S. No. | Other Protein Kinases (PDB ID) | MDPI (molMall) ID | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 21830 | 11352 | 15938 | 19037 | 21878 | 9539 | ||||||||
| Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | Score | ΔGbind | ||
| 1. | DYRK1B (4D2S) | −3.06 | −24.93 | −4.41 | −26.68 | −4.92 | −32.29 | −1.92 | −24.55 | −3.67 | −26.07 | −2.38 | −24.76 |
| 2. | DYRK2 (3KVW) | −4.41 | −28.27 | −4.82 | −35.88 | −4.48 | −38.08 | −3.33 | −26.51 | −4.78 | −29.17 | −1.91 | −29.86 |
| 3. | CDK5/p25 (3O0G) | −4.66 | −24.58 | −4.13 | −21.07 | −4.19 | −27.05 | −3.56 | −24.69 | −2.91 | −26.82 | −2.20 | −21.21 |
| 4. | CK1 (5W4W) | −4.32 | −24.40 | −3.56 | −23.85 | −1.61 | −28.72 | −3.31 | −18.65 | −1.91 | −17.94 | −3.29 | −10.20 |
| 5. | CLK1 (1Z57) | −3.21 | −24.05 | −3.25 | −25.85 | −2.04 | −31.94 | −3.32 | −24.49 | −4.52 | −24.03 | −1.75 | −21.87 |
| 6. | CLK3 (3RAW) | −3.96 | −22.60 | −4.72 | −29.45 | −3.60 | −28.74 | −3.34 | −26.27 | −2.75 | −30.81 | −4.04 | −21.52 |
| 7. | GSK3β (5OY4) | −3.54 | −27.03 | −4.47 | −27.37 | −1.90 | −34.49 | −3.72 | −27.34 | −2.36 | −23.14 | −4.03 | −29.32 |
| 8. | MAPK2 (2PZY) | −2.68 | −23.23 | −4.26 | −19.91 | −2.90 | −13.67 | −2.01 | −17.35 | −4.20 | −26.60 | −1.86 | −18.25 |
| 9. | MAPK10 (3RTP) | −4.17 | −25.13 | −4.06 | −26.56 | −2.97 | −37.01 | −2.92 | −18.56 | −3.64 | −20.31 | −2.69 | −29.62 |
| 10. | PIM1 (5VUA) | −2.47 | −29.84 | −3.07 | −19.72 | −2.46 | −29.90 | −2.38 | −27.41 | −2.65 | −26.85 | −2.86 | −27.67 |
| 11. | PKA (5LCP) | −2.38 | −26.41 | −4.46 | −21.17 | −3.84 | −21.99 | −1.79 | −23.15 | −4.50 | −25.08 | −0.70 | −19.48 |
| 12. | PKCα (1DSY) | −0.34 | −23.23 | −3.61 | −20.32 | −0.37 | −18.27 | −2.52 | −21.98 | −2.22 | −17.99 | 0.43 | −10.94 |
| molMall ID | Molecule SMILES | SA Score |
|---|---|---|
| 21830 | c1c2c(ccc1)c1c([nH]2)C(=O)N(CC1)CCc1ccc(cc1)OC | 70.672 |
| 11352 | c1c2c(cc3c1NC(=O)C3)/C(=C(\c1ccccc1)/C)/C(=O)N2 | 69.201 |
| 15938 | C1(=O)[C@@H]2[C@H](C(=O)[C@@H]3[C@H]1C=CC=C3)C=CC(=C2NC(=O)c1ccncc1)C | 44.679 |
| 19037 | C1(=O)/C(=C/Nc2ccccc2)/C(=O)N(N1)c1ccccc1 | 79.648 |
| 21878 | c1c2c(cc(c1)C(=O)OCC)c1c([nH]2)C(=O)OCC1 | 72.346 |
| 9539 | c1c2c(ccc1)n(c(=O)c1c2[nH]c(=O)[nH]nc1C)c1ccccc1 | 71.309 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahroz, M.M.; Sharma, H.K.; Altamimi, A.S.A.; Alamri, M.A.; Ali, A.; Ali, A.; Alqahtani, S.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; et al. Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study. Molecules 2022, 27, 1159. https://doi.org/10.3390/molecules27041159
Shahroz MM, Sharma HK, Altamimi ASA, Alamri MA, Ali A, Ali A, Alqahtani S, Altharawi A, Alabbas AB, Alossaimi MA, et al. Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study. Molecules. 2022; 27(4):1159. https://doi.org/10.3390/molecules27041159
Chicago/Turabian StyleShahroz, Mir Mohammad, Hemant Kumar Sharma, Abdulmalik S. A. Altamimi, Mubarak A. Alamri, Abuzer Ali, Amena Ali, Safar Alqahtani, Ali Altharawi, Alhumaidi B. Alabbas, Manal A. Alossaimi, and et al. 2022. "Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study" Molecules 27, no. 4: 1159. https://doi.org/10.3390/molecules27041159