
Part II: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Material Synthesis and Electrode-Fabrication Parameters on the Electrocatalytic Activity in the Hydrogen Evolution Reaction




Abstract
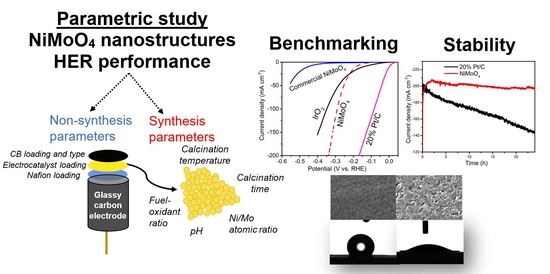
:
1. Introduction
2. Results and Discussion
2.1. Non-Synthesis Parameters
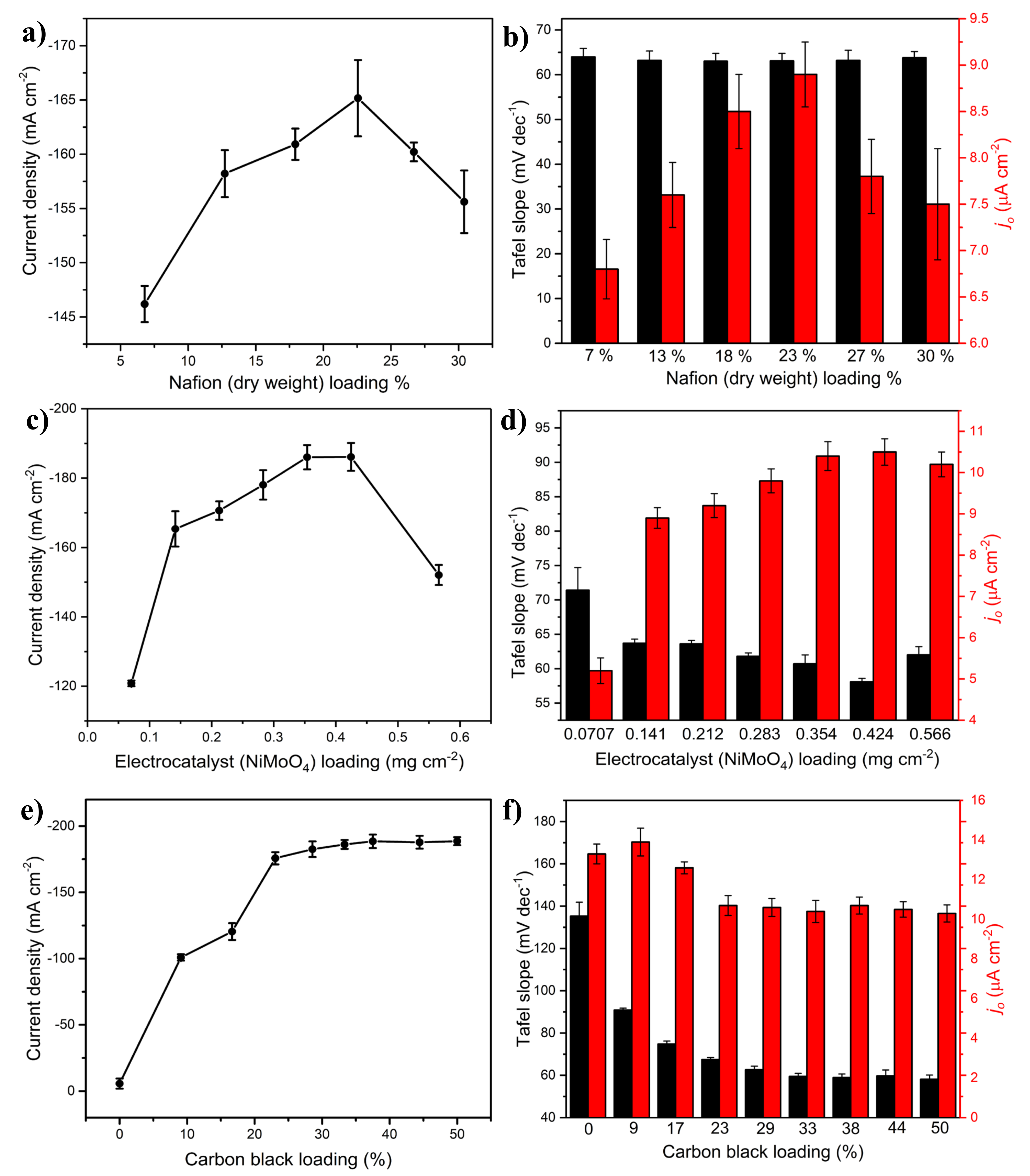
2.1.1. Effect of Nafion Loading
2.1.2. Effect of Catalyst (NiMoO4) and Carbon Black Loading
2.1.3. Effect of Carbon Black Type

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | BET * Surface Area (m2/g) | BET Surface Area of NiMoO4/CB Mixture with 9 wt.% CB (m2/g) | Pore Volume NiMoO4/CB Mixture with 9 wt.% CB (cm3/g) | OAN ** | Conductivity Index |
|---|---|---|---|---|---|
| XC-72R | 234 | 61.6 | 0.182 | 175 | 120 |
| Black Pearls 2000 | 1580 | 332.3 | 0.682 | 330 | 146 |
| LITX 200 | 159 | 42.9 | 0.154 | 162 | 21 |
| PBX 51 | 1420 | 237.2 | 0.275 | 170 | 119 |
| XCmax 22 | 1360 | 303.7 | 0.525 | 320 | 142 |
2.2. Synthesis Parameters
2.2.1. Effect of Calcination Temperature and Time
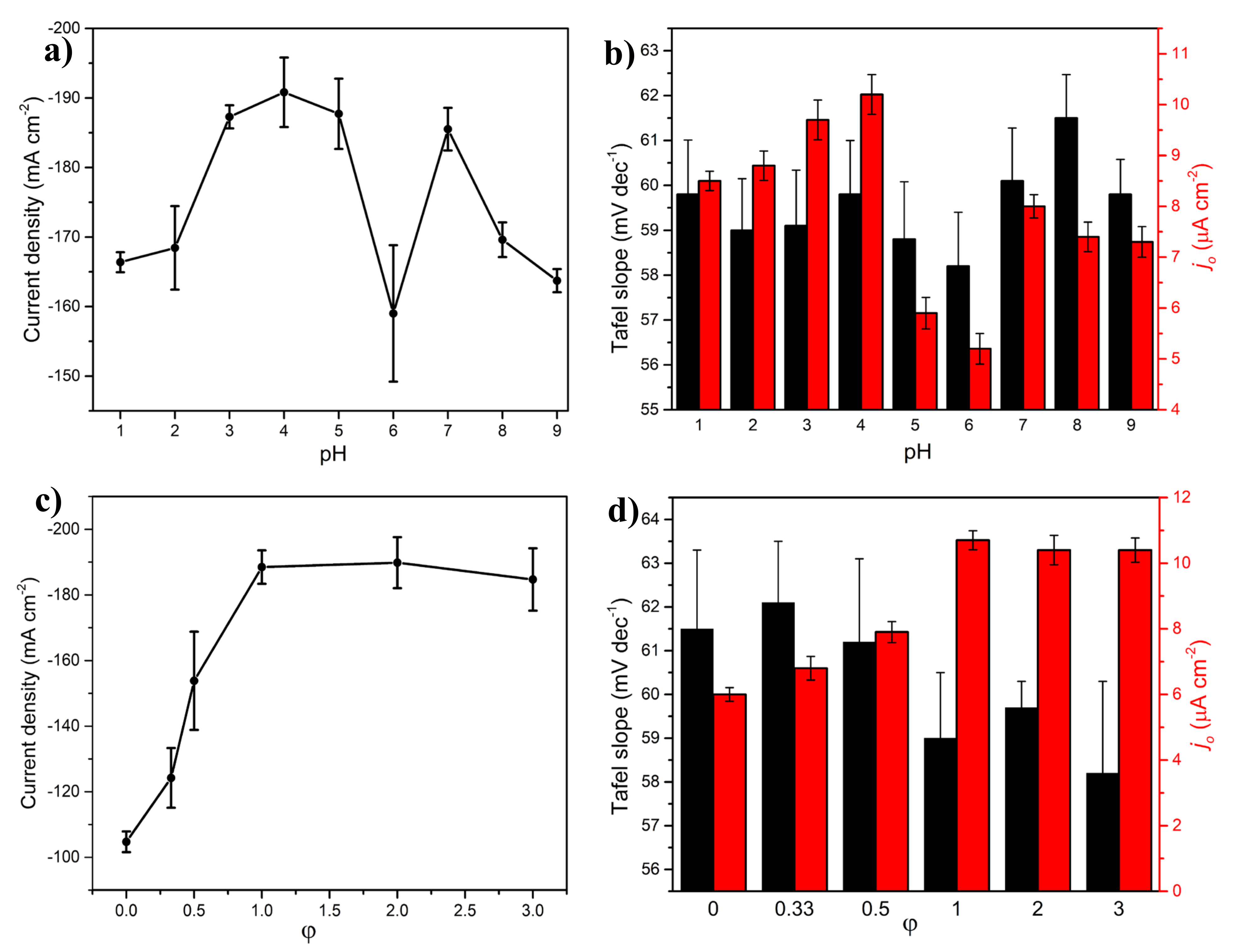
2.2.2. Effect of Ni-Mo-Oxide Composition
2.2.3. Effect of pH
2.2.4. Effect of Fuel Content
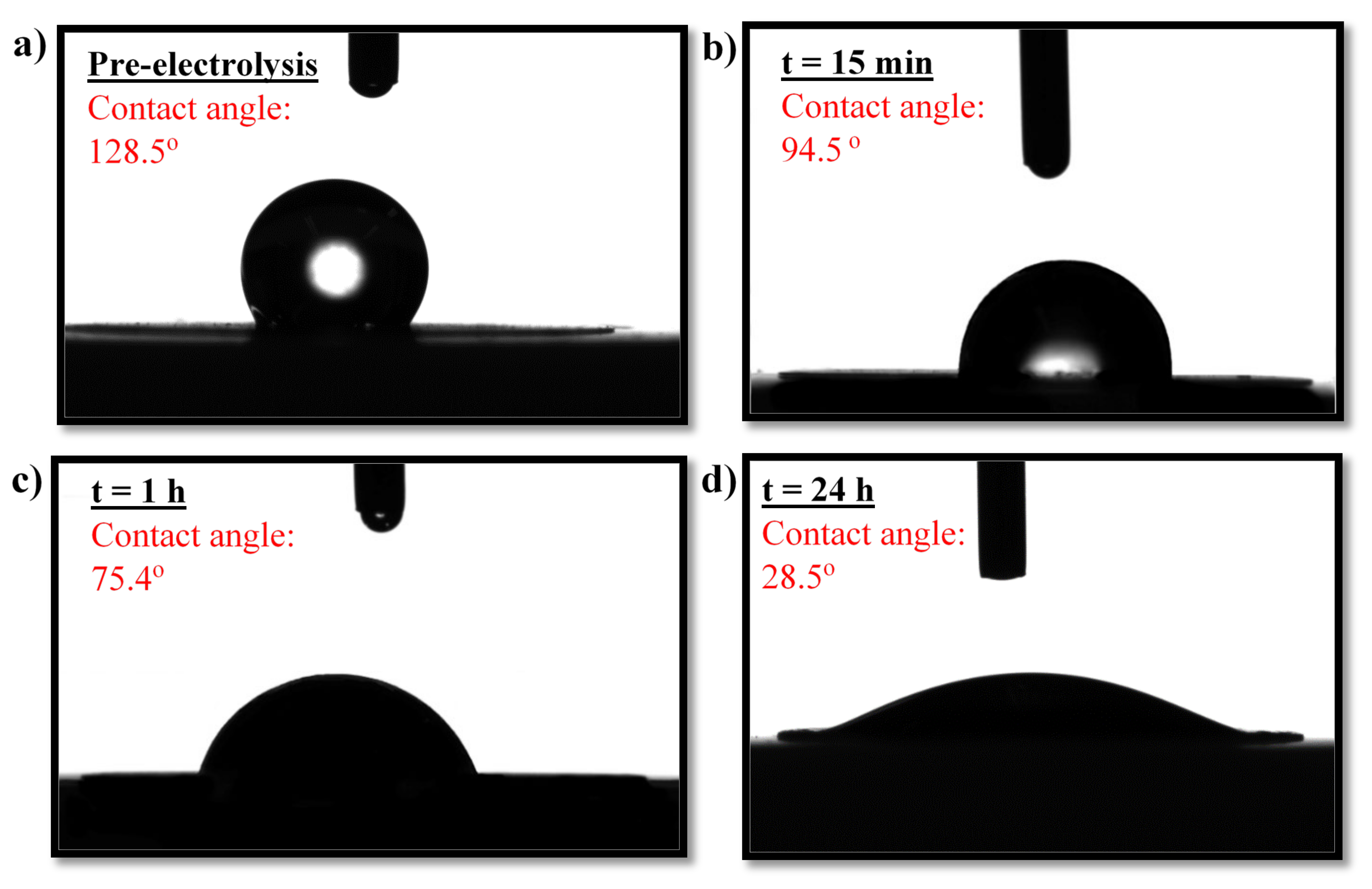
2.3. Benchmarking and Electrocatalyst Layer Stability
3. Materials and Methods
3.1. Sample Preparation
3.2. Electrochemical Characterization
3.3. Characterization Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE 2011, 100, 410–426. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Gong, X.; Guo, Z. The intensification technologies to water electrolysis for hydrogen production—A review. Renew. Sustain. Energy Rev. 2014, 29, 573–588. [Google Scholar] [CrossRef]
- Dyer, C.K. Fuel cells for portable applications. J. Power Sources 2002, 106, 31–34. [Google Scholar] [CrossRef]
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Stern, L.-A.; Hu, X. Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution. Chem. Soc. Rev. 2014, 43, 6555–6569. [Google Scholar] [CrossRef] [Green Version]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Barbir, F. PEM electrolysis for production of hydrogen from renewable energy sources. Sol. Energy 2005, 78, 661–669. [Google Scholar] [CrossRef]
- Nikolic, V.M.; Tasic, G.S.; Maksic, A.D.; Saponjic, D.P.; Miulovic, S.M.; Kaninski, M.P.M. Raising efficiency of hydrogen generation from alkaline water electrolysis–Energy saving. Int. J. Hydrogen Energy 2010, 35, 12369–12373. [Google Scholar] [CrossRef]
- Faber, M.S.; Jin, S. Earth-abundant inorganic electrocatalysts and their nanostructures for energy conversion applications. Energy Environ. Sci. 2014, 7, 3519–3542. [Google Scholar] [CrossRef]
- Kumar, S.S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni (OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Li, Y. Recent advances in heterogeneous electrocatalysts for the hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 14942–14962. [Google Scholar] [CrossRef]
- Navarro-Flores, E.; Chong, Z.; Omanovic, S. Characterization of Ni, NiMo, NiW and NiFe electroactive coatings as electrocatalysts for hydrogen evolution in an acidic medium. J. Mol. Catal. A Chem. 2005, 226, 179–197. [Google Scholar] [CrossRef]
- McKone, J.R.; Sadtler, B.F.; Werlang, C.A.; Lewis, N.S.; Gray, H.B. Ni–Mo nanopowders for efficient electrochemical hydrogen evolution. ACS Catal. 2013, 3, 166–169. [Google Scholar] [CrossRef]
- Yuan, M.; Zhu, Y.; Deng, L.; Ming, R.; Zhang, A.; Li, W.; Chai, B.; Ren, Z. IrO2–TiO2 electrocatalysts for the hydrogen evolution reaction in acidic water electrolysis without activation. New J. Chem. 2017, 41, 6152–6159. [Google Scholar] [CrossRef]
- Vidales, A.G.; Omanovic, S. Evaluation of nickel-molybdenum-oxides as cathodes for hydrogen evolution by water electrolysis in acidic, alkaline, and neutral media. Electrochim. Acta 2018, 262, 115–123. [Google Scholar] [CrossRef]
- Rammal, M.B.; Omanovic, S. Synthesis and characterization of NiO, MoO3, and NiMoO4 nanostructures through a green, facile method and their potential use as electrocatalysts for water splitting. Mater. Chem. Phys. 2020, 255, 123570. [Google Scholar] [CrossRef]
- Rammal, M.B.; Omanovic, S. Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties. Molecules 2022, 27, 776. [Google Scholar] [CrossRef]
- Peighambardoust, S.J.; Rowshanzamir, S.; Amjadi, M. Review of the proton exchange membranes for fuel cell applications. Int. J. Hydrogen Energy 2010, 35, 9349–9384. [Google Scholar] [CrossRef]
- Xue, N.; Diao, P. Molybdenum diselenide nanolayers prepared on carbon black as an efficient and stable electrocatalyst for hydrogen evolution reaction. J. Phys. Chem. 2017, 121, 26686–26697. [Google Scholar] [CrossRef]
- Cao, P.; Peng, J.; Li, J.; Zhai, M. Highly conductive carbon black supported amorphous molybdenum disulfide for efficient hydrogen evolution reaction. J. Power Sources 2017, 347, 210–219. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, S.; Pastor, E.; Lázaro, M. Electrochemical behavior of the carbon black Vulcan XC-72R: Influence of the surface chemistry. Int. J. Hydrogen Energy 2018, 43, 7911–7922. [Google Scholar] [CrossRef] [Green Version]
- Sanders, I.J.; Peeten, T.L. Carbon Black: Production, Properties and Uses; Nova Science Publishers: Hauppauge, NY, USA, 2011. [Google Scholar]
- Madeira, L.; Portela, M.; Mazzocchia, C. Nickel molybdate catalysts and their use in the selective oxidation of hydrocarbons. Catal. Rev. 2004, 46, 53–110. [Google Scholar] [CrossRef]
- Islam, I.; Sultana, S.; Kumer Ray, S.; Parvin Nur, H.; Hossain, M. Electrical and tensile properties of carbon black reinforced polyvinyl chloride conductive composites. C J. Carbon Res. 2018, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Dominko, R.; Gaberscek, M.; Drofenik, J.; Bele, M.; Pejovnik, S.; Jamnik, J. The role of carbon black distribution in cathodes for Li ion batteries. J. Power Sources 2003, 119, 770–773. [Google Scholar] [CrossRef]
- Zhang, Y.; Zang, J.; Han, C.; Jia, S.; Tian, P.; Gao, H.; Wang, Y. Molybdenum oxide and molybdenum carbide coated carbon black as an electrocatalyst for hydrogen evolution reaction in acidic media. Int. J. Hydrogen Energy 2017, 42, 26985–26994. [Google Scholar] [CrossRef]
- Khodabakhshi, S.; Fulvio, P.F.; Andreoli, E. Carbon black reborn: Structure and chemistry for renewable energy harnessing. Carbon 2020, 162, 604–649. [Google Scholar] [CrossRef] [Green Version]
- Joshi, U.; Lee, J.; Giordano, C.; Malkhandi, S.; Yeo, B.S. Enhanced catalysis of the electrochemical hydrogen evolution reaction using composites of molybdenum-based compounds, gold nanoparticles and carbon. Phys. Chem. Chem. Phys. 2016, 18, 21548–21553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafeeyan, M.S.; Daud, W.M.A.W.; Houshmand, A.; Shamiri, A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal. Appl. Pyrolysis 2010, 89, 143–151. [Google Scholar] [CrossRef]
- Carmo, M.; Dos Santos, A.R.; Poco, J.G.; Linardi, M. Physical and electrochemical evaluation of commercial carbon black as electrocatalysts supports for DMFC applications. J. Power Sources 2007, 173, 860–866. [Google Scholar] [CrossRef]
- Zhuang, X.-G.; Cheng, J.; Yang, D.-P.; Cao, G.-P.; Yang, Y.-S. Structure and electrochemical characters of nano-sized Ni(OH)2-C prepared by precipitation-transformation method. J. Inorg. Mater. 2005, 20, 337–344. [Google Scholar]
- Spahr, M.E.; Gilardi, R.; Bonacchi, D. Carbon black for electrically conductive polymer applications. In Fillers for Polymer Applications; Springer: Cham, Switzerland, 2016; p. 375. [Google Scholar]
- Čolić, V.; Yang, S.; Révay, Z.; Stephens, I.E.; Chorkendorff, I. Carbon catalysts for electrochemical hydrogen peroxide production in acidic media. Electrochim. Acta 2018, 272, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Wang, C.-W.; Liu, G.; Song, X.-Y.; Battaglia, V.; Sastry, A.M. Selection of conductive additives in li-ion battery cathodes: A numerical study. J. Electrochem. Soc. 2007, 154, A978. [Google Scholar] [CrossRef] [Green Version]
- Shang, H.; Lu, Y.; Zhao, F.; Chao, C.; Zhang, B.; Zhang, H. Preparing high surface area porous carbon from biomass by carbonization in a molten salt medium. RSC Adv. 2015, 5, 75728–75734. [Google Scholar] [CrossRef]
- Pantea, D.; Darmstadt, H.; Kaliaguine, S.; Roy, C. Electrical conductivity of conductive carbon blacks: Influence of surface chemistry and topology. Appl. Surf. Sci. 2003, 217, 181–193. [Google Scholar] [CrossRef]
- Pantea, D.; Darmstadt, H.; Kaliaguine, S.; Sümmchen, L.; Roy, C. Electrical conductivity of thermal carbon blacks: Influence of surface chemistry. Carbon 2001, 39, 1147–1158. [Google Scholar] [CrossRef]
- Assumpção, M.; De Souza, R.; Rascio, D.; Silva, J.; Calegaro, M.; Gaubeur, I.; Paixão, T.; Hammer, P.; Lanza, M.; Santos, M.C. A comparative study of the electrogeneration of hydrogen peroxide using Vulcan and Printex carbon supports. Carbon 2011, 49, 2842–2851. [Google Scholar] [CrossRef]
- Farquhar, A.K.; Supur, M.; Smith, S.R.; Van Dyck, C.; McCreery, R.L. Hybrid Graphene Ribbon/Carbon Electrodes for High-Performance Energy Storage. Adv. Energy Mater. 2018, 8, 1802439. [Google Scholar] [CrossRef]
- Korchev, A.; DuPasquier, A.; Atanassova, P. Oxidized Carbon Blacks and Applications for Lead Acid Batteries. U.S. Patent 9,923,205, 20 March 2018. [Google Scholar]
- Wang, F.; Gao, X.; Lu, Z.; Ye, S.; Qu, J.; Wu, F.; Yuan, H.; Song, D. Electrochemical properties of Mg-based alloys containing carbon nanotubes. J. Alloys Compd. 2004, 370, 326–330. [Google Scholar] [CrossRef]
- Youssry, M.; Kamand, F.Z.; Magzoub, M.I.; Nasser, M.S. Aqueous dispersions of carbon black and its hybrid with carbon nanofibers. RSC Adv. 2018, 8, 32119–32131. [Google Scholar] [CrossRef] [Green Version]
- Amadine, O.; Essamlali, Y.; Fihri, A.; Larzek, M.; Zahouily, M. Effect of calcination temperature on the structure and catalytic performance of copper–ceria mixed oxide catalysts in phenol hydroxylation. RSC Adv. 2017, 7, 12586–12597. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Sk, M.A.; Thia, L.; Ge, X.; Lim, R.J.; Wang, J.-Y.; Lim, K.H.; Wang, X. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ. Sci. 2014, 7, 2624–2629. [Google Scholar] [CrossRef] [Green Version]
- Pillay, B.; Mathebula, M.R.; Friedrich, H.B. The oxidative dehydrogenation of n-hexane over Ni–Mo–O catalysts. Appl. Catal. A Gen. 2009, 361, 57–64. [Google Scholar] [CrossRef]
- Ratha, S.; Samantara, A.K.; Singha, K.K.; Gangan, A.S.; Chakraborty, B.; Jena, B.K.; Rout, C.S. Urea-assisted room temperature stabilized metastable β-NiMoO4: Experimental and theoretical insights into its unique bifunctional activity toward oxygen evolution and supercapacitor. ACS Appl. Mater. Interfaces 2017, 9, 9640–9653. [Google Scholar] [CrossRef]
- Mazzocchia, C.; Aboumrad, C.; Diagne, C.; Tempesti, E.; Herrmann, J.; Thomas, G. On the NiMoO4 oxidative dehydrogenation of propane to propene: Some physical correlations with the catalytic activity. Catal. Lett. 1991, 10, 181–191. [Google Scholar] [CrossRef]
- Chen, W.F.; Sasaki, K.; Ma, C.; Frenkel, A.I.; Marinkovic, N.; Muckerman, J.T.; Zhu, Y.; Adzic, R.R. Hydrogen-evolution catalysts based on non-noble metal nickel—Molybdenum nitride nanosheets. Angew. Chem. Int. Ed. 2012, 51, 6131–6135. [Google Scholar] [CrossRef]
- Damian, A.; Omanovic, S. Ni and NiMo hydrogen evolution electrocatalysts electrodeposited in a polyaniline matrix. J. Power Sources 2006, 158, 464–476. [Google Scholar] [CrossRef]
- Yue, Z.; Guo, W.; Zhou, J.; Gui, Z.; Li, L. Synthesis of nanocrystilline ferrites by sol–gel combustion process: The influence of pH value of solution. J. Magn. Magn. Mater. 2004, 270, 216–223. [Google Scholar] [CrossRef]
- Chen, Z.; Cummins, D.; Reinecke, B.N.; Clark, E.; Sunkara, M.K.; Jaramillo, T.F. Core–shell MoO3–MoS2 nanowires for hydrogen evolution: A functional design for electrocatalytic materials. Nano Lett. 2011, 11, 4168–4175. [Google Scholar] [CrossRef]
- Deganello, F.; Tyagi, A.K. Solution combustion synthesis, energy and environment: Best parameters for better materials. Prog. Cryst. Growth Charact. Mater. 2018, 64, 23–61. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Deng, L.; Li, W.; Ren, Z.; Yang, M.; Yang, X.; Zhu, Y. Synthesis and characterization of an IrO2–Fe2O3 electrocatalyst for the hydrogen evolution reaction in acidic water electrolysis. RSC Adv. 2017, 7, 20252–20258. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Han, X.; Gadipelli, S.; Guo, J.; Wu, S.; Kang, L.; Callison, J.; Guo, Z. In situ synthesized low-PtCo@ porous carbon catalyst for highly efficient hydrogen evolution. J. Mater. Chem. A 2019, 7, 6543–6551. [Google Scholar] [CrossRef] [Green Version]
- Bessarabov, D.; Millet, P. PEM Water Electrolysis; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Kötz, E.; Stucki, S. Ruthenium dioxide as a hydrogen-evolving cathode. J. Appl. Electrochem. 1987, 17, 1190–1197. [Google Scholar] [CrossRef]
- Wei, R.; Fang, M.; Dong, G.; Ho, J.C. Is platinum a suitable counter electrode material for electrochemical hydrogen evolution reaction? Sci. Bull. 2017, 62, 971–973. [Google Scholar] [CrossRef] [Green Version]
- Blouin, M.; Guay, D. Activation of ruthenium oxide, iridium oxide, and mixed RuxIr1-x oxide electrodes during cathodic polarization and hydrogen evolution. J. Electrochem. Soc. 1997, 144, 573. [Google Scholar] [CrossRef]
- Chen, L.; Guay, D.; Pollak, F.; Levy, F. AFM observation of surface activation of ruthenium oxide electrodes during hydrogen evolution. J. Electroanal. Chem. 1997, 429, 185–192. [Google Scholar] [CrossRef]
- Yi, Y.; Weinberg, G.; Prenzel, M.; Greiner, M.; Heumann, S.; Becker, S.; Schlögl, R. Electrochemical corrosion of a glassy carbon electrode. Catal. Today 2017, 295, 32–40. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, H.; Hu, Y.; Jiang, H.; Li, C. Integrated Ni-P-S nanosheets array as superior electrocatalysts for hydrogen generation. Green Energy Environ. 2017, 2, 112–118. [Google Scholar] [CrossRef]
- Goswami, S.; Klaus, S.; Benziger, J. Wetting and absorption of water drops on Nafion films. Langmuir 2008, 24, 8627–8633. [Google Scholar] [CrossRef]
- Curtin, D.E.; Lousenberg, R.D.; Henry, T.J.; Tangeman, P.C.; Tisack, M.E. Advanced materials for improved PEMFC performance and life. J. Power Sources 2004, 131, 41–48. [Google Scholar] [CrossRef]
- Jiang, Y.; Hao, J.; Hou, M.; Zhang, H.; Li, X.; Shao, Z.; Yi, B. A new microporous layer material to improve the performance and durability of polymer electrolyte membrane fuel cells. RSC Adv. 2015, 5, 104095–104100. [Google Scholar] [CrossRef]
- Santiago, D.; Rodríguez-Calero, G.G.; Rivera, H.; Tryk, D.A.; Scibioh, M.A.; Cabrera, C.R. Platinum electrodeposition at high surface area carbon vulcan-XC-72R material using a rotating disk-slurry electrode technique. J. Electrochem. Soc. 2010, 157, F189. [Google Scholar] [CrossRef]
- Rosales-Leal, J.; Rodríguez-Valverde, M.; Mazzaglia, G.; Ramón-Torregrosa, P.; Díaz-Rodríguez, L.; García-Martínez, O.; Vallecillo-Capilla, M.; Ruiz, C.; Cabrerizo-Vílchez, M. Effect of roughness, wettability and morphology of engineered titanium surfaces on osteoblast-like cell adhesion. Colloids Surf. A Physicochem. Eng. Asp. 2010, 365, 222–229. [Google Scholar] [CrossRef]
- Chabanier, C.; Guay, D. Activation and hydrogen absorption in thermally prepared RuO2 and IrO2. J. Electroanal. Chem. 2004, 570, 13–27. [Google Scholar] [CrossRef]
- Chavan, H.S.; Hou, B.; Ahmed, A.T.A.; Kim, J.; Jo, Y.; Cho, S.; Park, Y.; Pawar, S.M.; Inamdar, A.I.; Cha, S.N. Ultrathin Ni-Mo oxide nanoflakes for high-performance supercapacitor electrodes. J. Alloys Compd. 2018, 767, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Chavan, H.S.; Hou, B.; Ahmed, A.T.A.; Jo, Y.; Cho, S.; Kim, J.; Pawar, S.M.; Cha, S.; Inamdar, A.I.; Im, H. Nanoflake NiMoO4 based smart supercapacitor for intelligent power balance monitoring. Sol. Energy Mater. Sol. Cells 2018, 185, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Haque, F.; Mohiuddin, M.; Carey, B.; Syed, N.; Zavabeti, A.; Zhang, B.; Khan, H.; Berean, K.; Ou, J. Highly active two-dimensional α-MoO3−x for the electrocatalytic hydrogen evolution reaction. J. Mater. Chem. A 2017, 5, 24223–24231. [Google Scholar] [CrossRef]
- Hoffmann, E.A.; Fekete, Z.A.; Korugic-Karasz, L.S.; Karasz, F.E.; Wilusz, E. Theoretical and experimental X-ray photoelectron spectroscopy investigation of ion-implanted nafion. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 551–556. [Google Scholar] [CrossRef]
- Friedman, A.K.; Shi, W.; Losovyj, Y.; Siedle, A.R.; Baker, L.A. Mapping microscale chemical heterogeneity in Nafion membranes with X-ray photoelectron spectroscopy. J. Electrochem. Soc. 2018, 165, H733. [Google Scholar] [CrossRef]
- Tyczkowski, J.; Balcerzak, J.; Sielski, J.; Krawczyk-Kłys, I. Effect of Carbon Black Nanofiller on Adhesion Properties of SBS Rubber Surfaces Treated by Low-Pressure Plasma. Polymers 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Murad, L.; Laffir, F.; Dickinson, C.; Dempsey, E. Pt based nanocomposites (mono/bi/tri-metallic) decorated using different carbon supports for methanol electro-oxidation in acidic and basic media. Nanoscale 2011, 3, 3334–3349. [Google Scholar] [CrossRef] [PubMed]





| Material | BET Surface Area (m2/g) | BJH Pore Volume (cm3/g) |
|---|---|---|
| NiMoO4 | 25.8 | 0.130 |
| XC-72R | 238 | 0.560 |
| 91% NiMoO4 + 9% XC-72R | 61.6 | 0.182 |
| 71% NiMoO4 + 29% XC-72R | 112.7 | 0.262 |
| Sample | Element (at.%) | ||
|---|---|---|---|
| Carbon | Oxygen | Sulfur | |
| XC-72R | 99.48 | 0.33 | 0.18 |
| Black Pearls 2000 | 97.01 | 2.65 | 0.34 |
| LITX 2000 | 94.89 | 4.09 | 0.18 |
| PBX 51 | 98.64 | 1.36 | * |
| XCmax 22 | 97.24 | 2.59 | 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rammal, M.B.; El-Ghoubaira, V.; Omanovic, S. Part II: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Material Synthesis and Electrode-Fabrication Parameters on the Electrocatalytic Activity in the Hydrogen Evolution Reaction. Molecules 2022, 27, 1199. https://doi.org/10.3390/molecules27041199
Rammal MB, El-Ghoubaira V, Omanovic S. Part II: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Material Synthesis and Electrode-Fabrication Parameters on the Electrocatalytic Activity in the Hydrogen Evolution Reaction. Molecules. 2022; 27(4):1199. https://doi.org/10.3390/molecules27041199
Chicago/Turabian StyleRammal, Mahmoud Bassam, Vincent El-Ghoubaira, and Sasha Omanovic. 2022. "Part II: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Material Synthesis and Electrode-Fabrication Parameters on the Electrocatalytic Activity in the Hydrogen Evolution Reaction" Molecules 27, no. 4: 1199. https://doi.org/10.3390/molecules27041199