
A Novel Small Molecular Prostaglandin Receptor EP4 Antagonist, L001, Suppresses Pancreatic Cancer Metastasis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
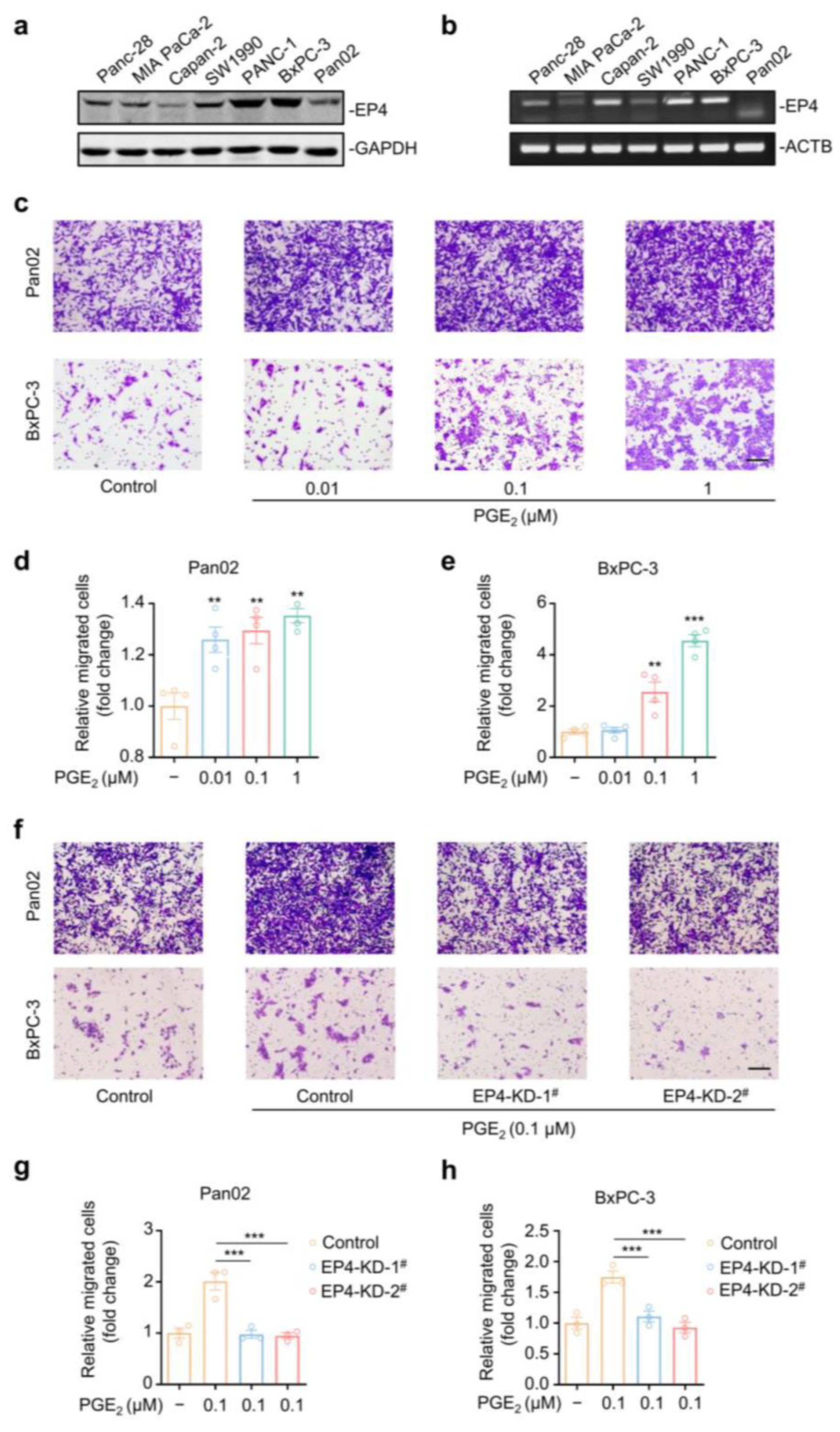
2.1. EP4 Is Essential for PGE2-Induced Pancreatic Cancer Cell Migration
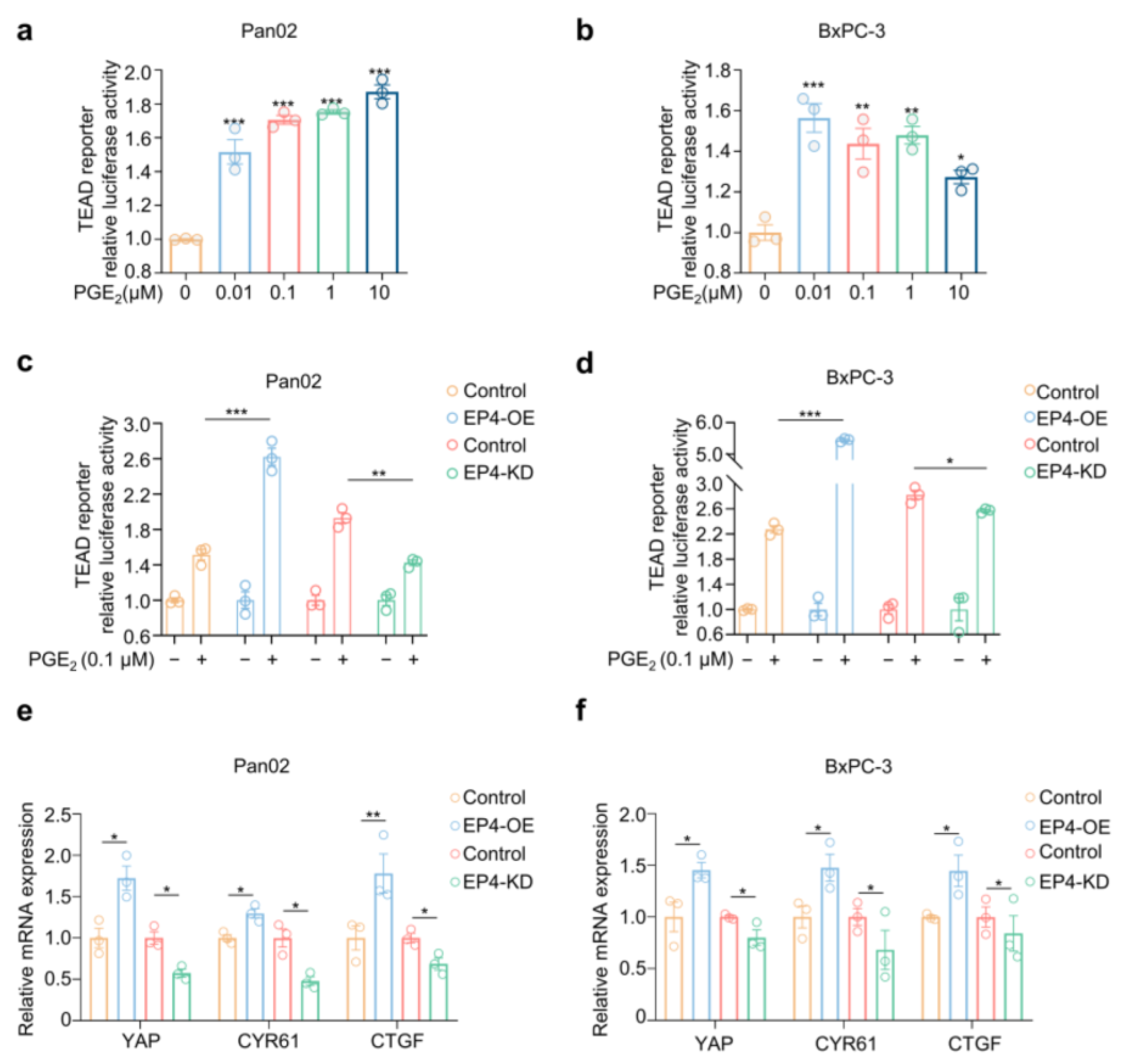
2.2. PGE2–EP4 Signaling Activates Hippo–Yap Pathway in Pancreatic Cancer Cells
2.3. L001 Is a Potent and Selective Antagonist of EP4 Receptor
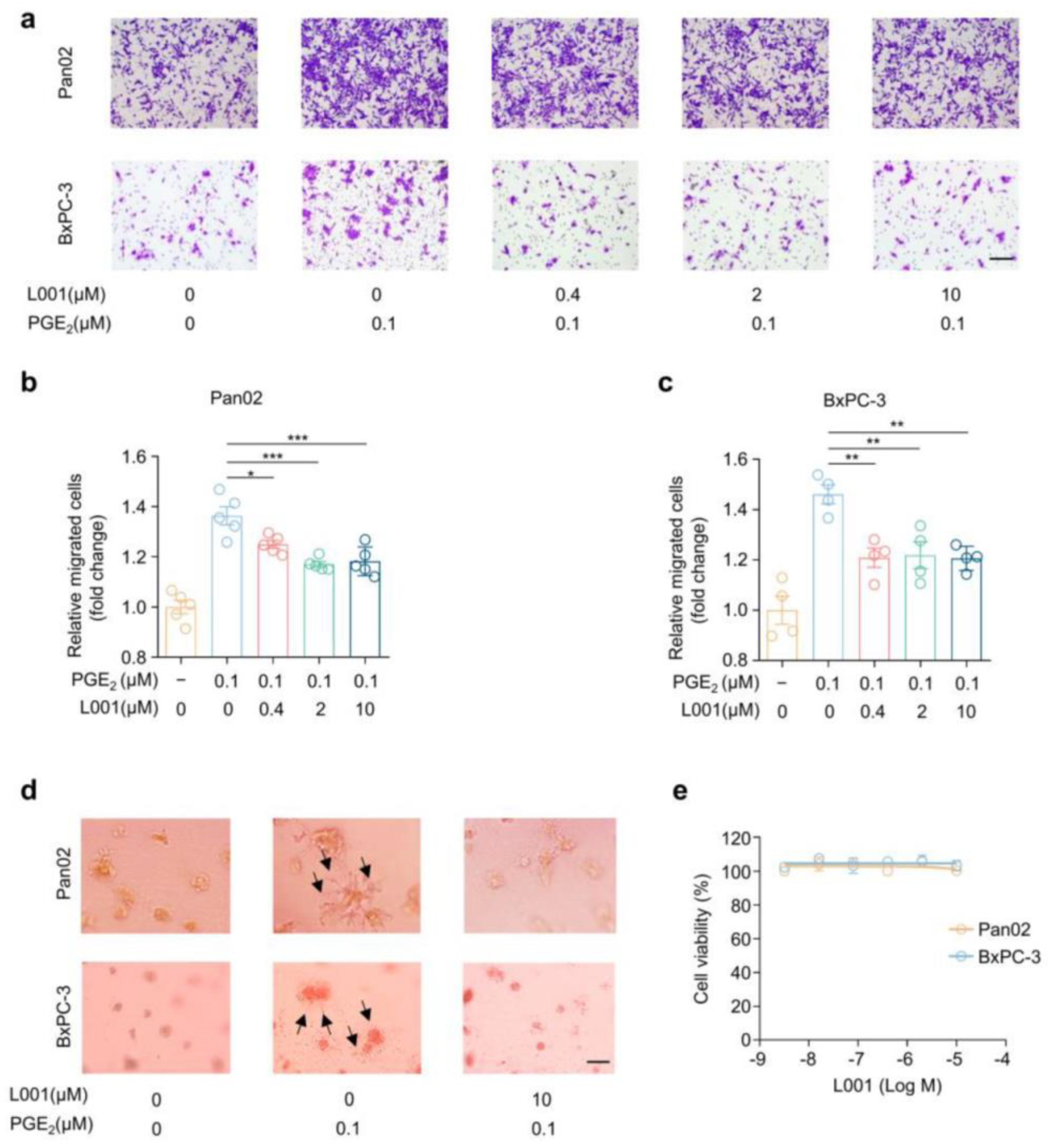
2.4. L001 Impairs Pancreatic Cancer Migration and Invasion In Vitro
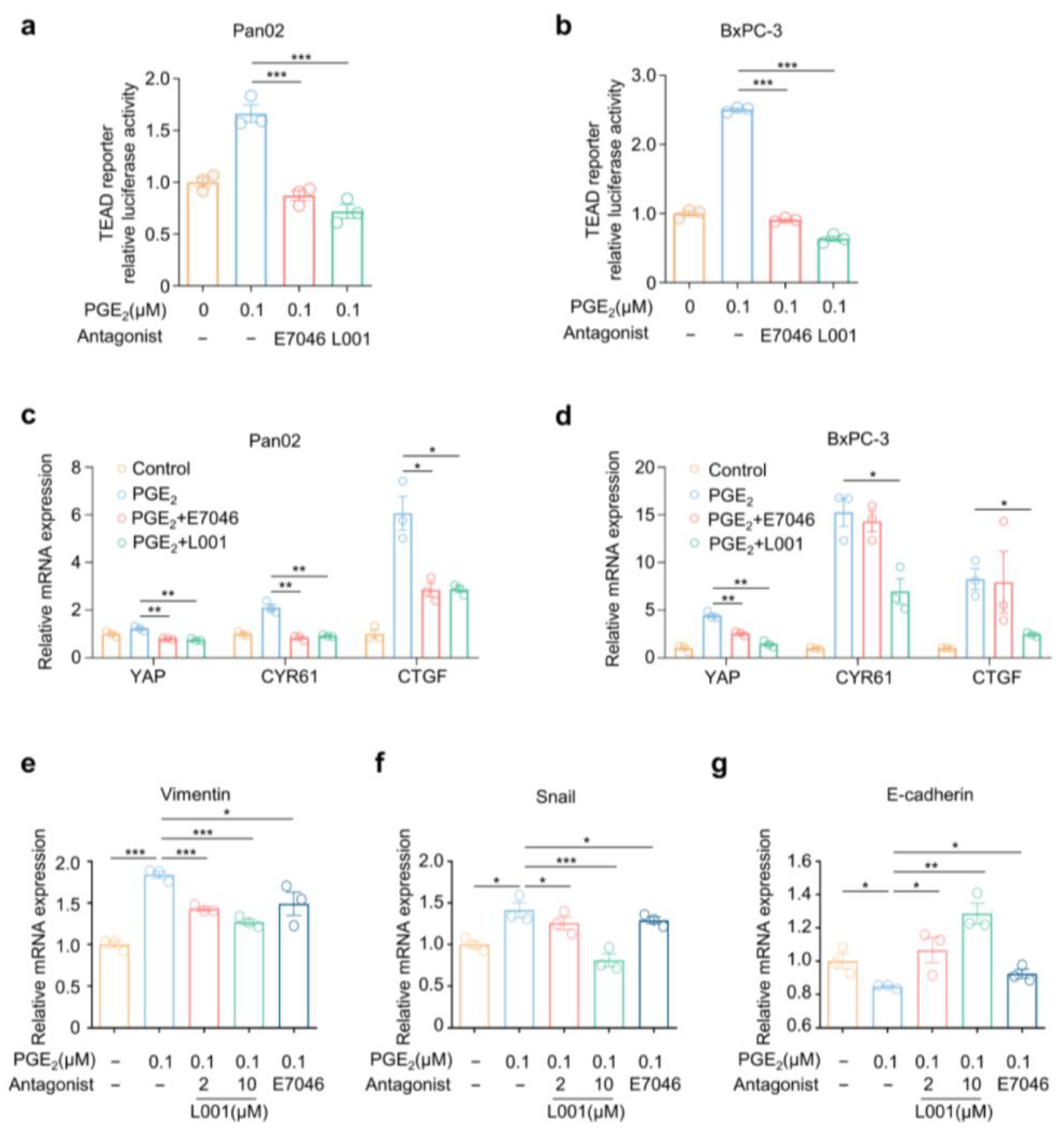
2.5. L001 Abrogated Hippo–YAP Activation and Pro-Metastatic Factors Expression in Pancreatic Cancer Cells
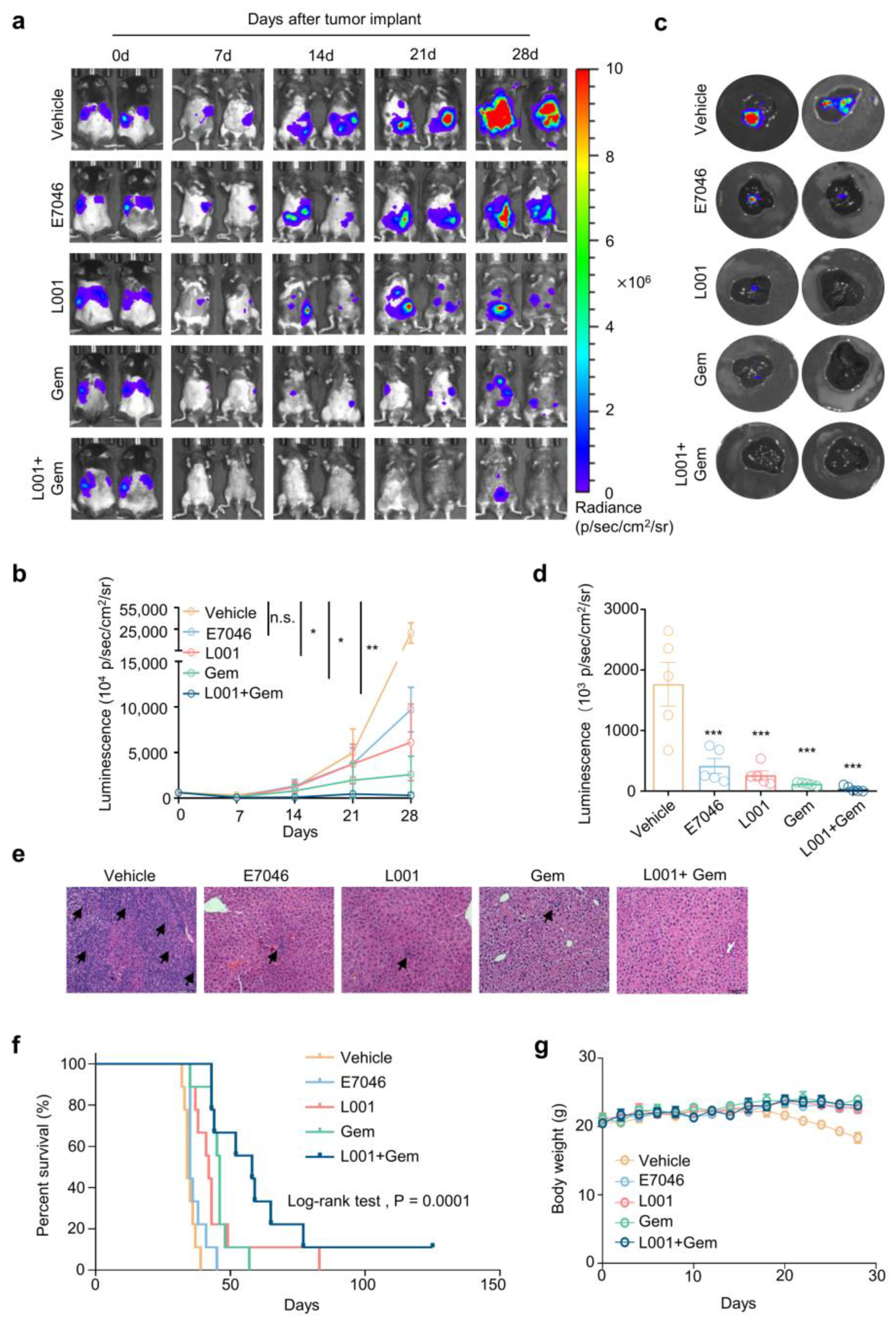
2.6. L001 Treatment Impairs Hepatic Metastasis of Pancreatic Cancer In Vivo
3. Discussion
4. Materials and Methods
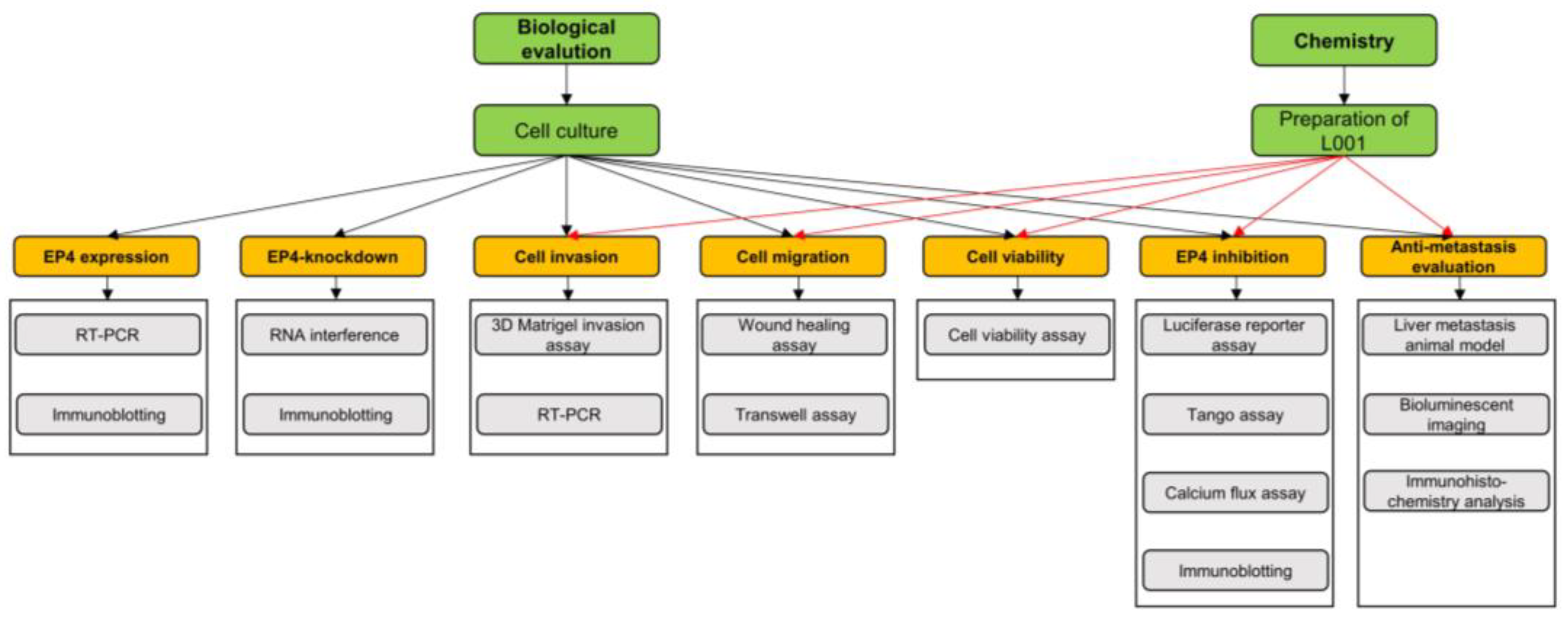
4.1. Experimental Framework
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Wound Healing Assay
4.5. Transwell Assay
4.6. 3D Matrigel Invasion Assay
4.7. Real-Time qPCR
4.8. Immunoblotting
4.9. RNA Interference
4.10. Luciferase Reporter Assay
4.11. TANGO Assay
4.12. Calcium Flux Assay
4.13. Liver Metastasis Animal Model and Bioluminescent Imaging
4.14. Immunohistochemistry Analysis
4.15. Chemistry
4.15.1. Preparation of L001
4.15.2. Reagents and Conditions
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Vaccaro, V.; Sperduti, I.; Milella, M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 365, 768–769. [Google Scholar]
- Jia, Y.; Gu, D.; Wan, J.; Yu, B.; Zhang, X.; Chiorean, E.G.; Wang, Y.; Xie, J. The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene 2019, 38, 1764–1777. [Google Scholar] [CrossRef] [Green Version]
- Lundy, J.; Gearing, L.J.; Gao, H.; West, A.C.; McLeod, L.; Deswaerte, V.; Yu, L.; Porazinski, S.; Pajic, M.; Hertzog, P.J.; et al. TLR2 activation promotes tumour growth and associates with patient survival and chemotherapy response in pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 6007–6022. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Role of prostanoids in gastrointestinal cancer. J. Clin. Investig. 2018, 128, 2732–2742. [Google Scholar] [CrossRef]
- Doherty, G.A.; Byrne, S.M.; Molloy, E.S.; Malhotra, V.; Austin, S.C.; Kay, E.W.; Murray, F.E.; Fitzgerald, D.J. Proneoplastic effects of PGE2 mediated by EP4 receptor in colorectal cancer. BMC Cancer 2009, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Xie, J.; Bedolla, R.; Rivas, P.; Chakravarthy, D.; Freeman, J.W.; Reddick, R.; Kopetz, S.; Peterson, A.; Wang, H.; et al. Combined targeting of STAT3/NF-kappaB/COX-2/EP4 for effective management of pancreatic cancer. Clin. Cancer Res. 2014, 20, 1259–1273. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Huang, Y.; Porta, R.; Yanagisawa, K.; Gonzalez, A.; Segi, E.; Johnson, D.H.; Narumiya, S.; Carbone, D.P. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006, 66, 9665–9672. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Charindra, D.; Shrestha, M.; Umehara, H.; Ogawa, I.; Miyauchi, M.; Takata, T. Tissue inhibitor of metalloproteinase-1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene 2018, 37, 263–270. [Google Scholar] [CrossRef]
- Mao, W.; Mai, J.; Peng, H.; Wan, J.; Sun, T. YAP in pancreatic cancer: Oncogenic role and therapeutic strategy. Theranostics 2021, 11, 1753–1762. [Google Scholar] [CrossRef]
- Hayashi, H.; Uemura, N.; Zhao, L.; Matsumura, K.; Sato, H.; Shiraishi, Y.; Baba, H. Biological Significance of YAP/TAZ in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 700315. [Google Scholar] [CrossRef]
- Cen, B.; Lang, J.D.; Du, Y.; Wei, J.; Xiong, Y.; Bradley, N.; Wang, D.; DuBois, R.N. Prostaglandin E2 Induces miR675-5p to Promote Colorectal Tumor Metastasis via Modulation of p53 Expression. Gastroenterology 2020, 158, 971–984. [Google Scholar] [CrossRef]
- Konya, V.; Marsche, G.; Schuligoi, R.; Heinemann, A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013, 138, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, C.Y.; Zha, Z.Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. Correction: TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2019, 294, 5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Lin, S.J.; Chen, Y.; Voon, D.C.; Zhu, F.; Chuang, L.S.; Wang, T.; Tan, P.; Lee, S.C.; Yeoh, K.G.; et al. RUNX3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene 2016, 35, 2664–2674. [Google Scholar] [CrossRef]
- Hawcroft, G.; Ko, C.W.; Hull, M.A. Prostaglandin E2-EP4 receptor signalling promotes tumorigenic behaviour of HT-29 human colorectal cancer cells. Oncogene 2007, 26, 3006–3019. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Parikh, A.; Shapiro, G.I.; Varga, A.; Naing, A.; Meric-Bernstam, F.; Ataman, O.; Reyderman, L.; Binder, T.A.; Ren, M.; et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 2020, 8, e000222. [Google Scholar] [CrossRef]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [Green Version]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.P.; Lansu, K.; McCorvy, J.D.; Giguere, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Ching, M.M.; Reader, J.; Fulton, A.M. Eicosanoids in Cancer: Prostaglandin E2 Receptor 4 in Cancer Therapeutics and Immunotherapy. Front. Pharmacol. 2020, 11, 819. [Google Scholar] [CrossRef]
- Lu, W.; Yu, W.; He, J.; Liu, W.; Yang, J.; Lin, X.; Zhang, Y.; Wang, X.; Jiang, W.; Luo, J.; et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol. Med. 2021, 13, e12798. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Kim, W.J.; Daaka, Y. PGE2 promotes renal carcinoma cell invasion through activated RalA. Oncogene 2013, 32, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhou, C.; Cheng, L.; Yan, B.; Chen, K.; Chen, X.; Zong, L.; Lei, J.; Duan, W.; Xu, Q.; et al. Inhibiting YAP expression suppresses pancreatic cancer progression by disrupting tumor-stromal interactions. J. Exp. Clin. Cancer Res. 2018, 37, 69. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Lillemoe, K.D.; Yeo, C.J.; Cameron, J.L. Pancreatic cancer: State-of-the-art care. CA Cancer J. Clin. 2000, 50, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, L.; Yuan, Z.; Lou, J.; Fan, Y.; Shi, A.; Huang, J.; Zhao, M.; Wu, Y. Multi-institutional development and external validation of machine learning-based models to predict relapse risk of pancreatic ductal adenocarcinoma after radical resection. J. Transl. Med. 2021, 19, 281. [Google Scholar] [CrossRef]
- Carmichael, J. Clinical response benefit in patients with advanced pancreatic cancer. Role of gemcitabine. Digestion 1997, 58, 503–507. [Google Scholar] [CrossRef]
- Jara-Gutierrez, A.; Baladron, V. The Role of Prostaglandins in Different Types of Cancer. Cells 2021, 10, 1487. [Google Scholar] [CrossRef]
- Papalazarou, V.; Zhang, T.; Paul, N.R.; Juin, A.; Cantini, M.; Maddocks, O.D.K.; Salmeron-Sanchez, M.; Machesky, L.M. The creatine-phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat. Metab. 2020, 2, 62–80. [Google Scholar] [CrossRef]
- Eibl, G.; Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (YAP) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal Transduct. Target Ther. 2018, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.J.; Yu, W.W.; Hu, L.L.; Liu, W.J.; Lin, X.H.; Wang, W.; Zhang, Q.; Wang, P.L.; Tang, S.W.; Wang, X.; et al. Discovery and Characterization of 1H-1,2,3-Triazole Derivatives as Novel Prostanoid EP4 Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem. 2020, 63, 569–590. [Google Scholar] [CrossRef]
- Albu, D.I.; Wang, Z.; Huang, K.C.; Wu, J.; Twine, N.; Leacu, S.; Ingersoll, C.; Parent, L.; Lee, W.; Liu, D.; et al. EP4 Antagonism by E7046 diminishes Myeloid immunosuppression and synergizes with Treg-reducing IL-2-Diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology 2017, 6, e1338239. [Google Scholar] [CrossRef]
- Nakao, K.; Murase, A.; Ohshiro, H.; Okumura, T.; Taniguchi, K.; Murata, Y.; Masuda, M.; Kato, T.; Okumura, Y.; Takada, J. CJ-023,423, a novel, potent and selective prostaglandin EP4 receptor antagonist with antihyperalgesic properties. J. Pharmacol. Exp. Ther. 2007, 322, 686–694. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef]
- Wright, J.J. Combination therapy of bortezomib with novel targeted agents: An emerging treatment strategy. Clin. Cancer Res. 2010, 16, 4094–4104. [Google Scholar] [CrossRef] [Green Version]
- De Dosso, S.; Siebenhuner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef]
- Dioufa, N.; Schally, A.V.; Chatzistamou, I.; Moustou, E.; Block, N.L.; Owens, G.K.; Papavassiliou, A.G.; Kiaris, H. Acceleration of wound healing by growth hormone-releasing hormone and its agonists. Proc. Natl. Acad. Sci. USA 2010, 107, 18611–18615. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, Y.; Shao, T.; Pei, H.; Guo, W.; Mi, D.; Krimm, I.; Zhang, Y.; Wang, P.; Wang, X.; et al. Modification and Biological Evaluation of a Series of 1,5-Diaryl-1,2,4-triazole Compounds as Novel Agents against Pancreatic Cancer Metastasis through Targeting Myoferlin. J. Med. Chem. 2019, 62, 4949–4966. [Google Scholar] [CrossRef]
- Tang, X.; Jin, R.; Qu, G.; Wang, X.; Li, Z.; Yuan, Z.; Zhao, C.; Siwko, S.; Shi, T.; Wang, P.; et al. GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Galphaq-p63RhoGEF-Rho GTPase pathway. Cancer Res. 2013, 73, 6206–6218. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, D.A.; Newton, R.; Zhu, Y.M.; El-Haroun, H.; Corbett, L.; Knox, A.J. Cyclooxygenase-2 induction by bradykinin in human pulmonary artery smooth muscle cells is mediated by the cyclic AMP response element through a novel autocrine loop involving endogenous prostaglandin E2, E-prostanoid 2 (EP2), and EP4 receptors. J. Biol. Chem. 2003, 278, 49954–49964. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell. Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Lin, X.; Meng, F.; Zhao, Y.; Wang, W.; Zhang, Y.; Chai, X.; Zhang, Y.; Yu, W.; Yang, J.; et al. A Novel Small Molecular Prostaglandin Receptor EP4 Antagonist, L001, Suppresses Pancreatic Cancer Metastasis. Molecules 2022, 27, 1209. https://doi.org/10.3390/molecules27041209
He J, Lin X, Meng F, Zhao Y, Wang W, Zhang Y, Chai X, Zhang Y, Yu W, Yang J, et al. A Novel Small Molecular Prostaglandin Receptor EP4 Antagonist, L001, Suppresses Pancreatic Cancer Metastasis. Molecules. 2022; 27(4):1209. https://doi.org/10.3390/molecules27041209
Chicago/Turabian StyleHe, Jiacheng, Xianhua Lin, Fanhui Meng, Yumiao Zhao, Wei Wang, Yao Zhang, Xiaolei Chai, Ying Zhang, Weiwei Yu, Junjie Yang, and et al. 2022. "A Novel Small Molecular Prostaglandin Receptor EP4 Antagonist, L001, Suppresses Pancreatic Cancer Metastasis" Molecules 27, no. 4: 1209. https://doi.org/10.3390/molecules27041209