
Design and Synthesis of Arylpiperazine Serotonergic/Dopaminergic Ligands with Neuroprotective Properties

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
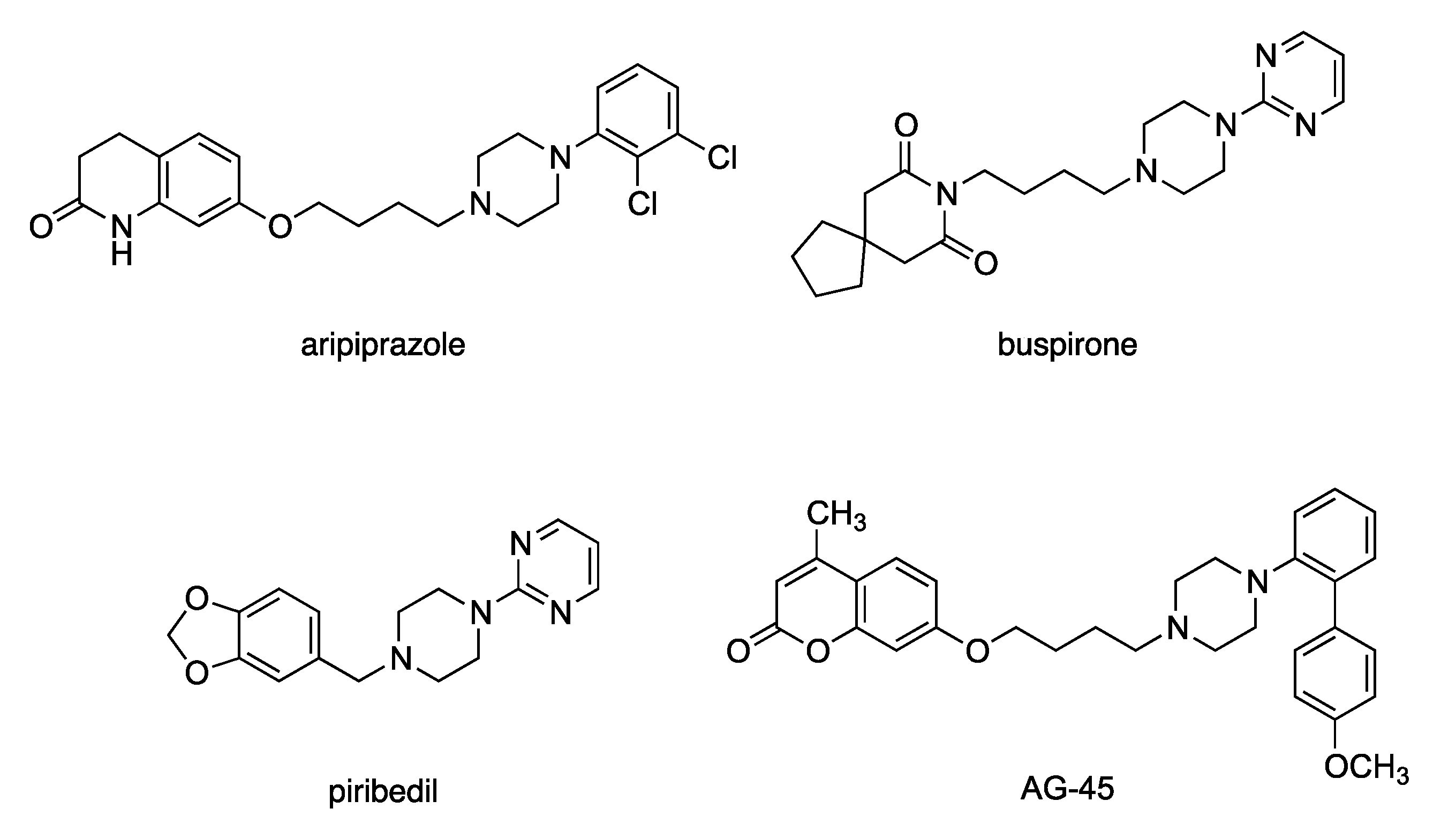
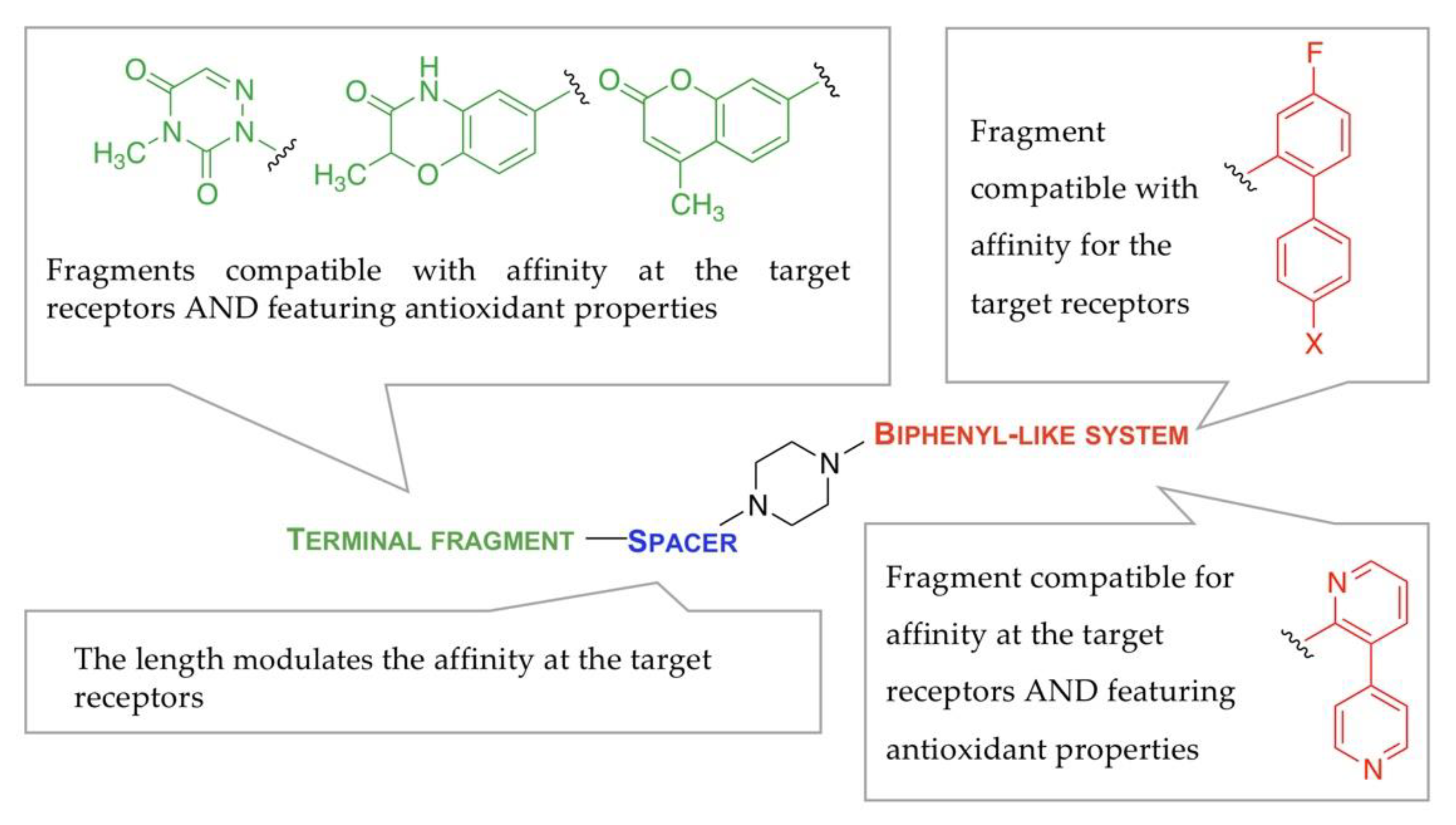
2.1. Study Design
2.2. Chemistry
2.3. Radioligand Binding Experiments
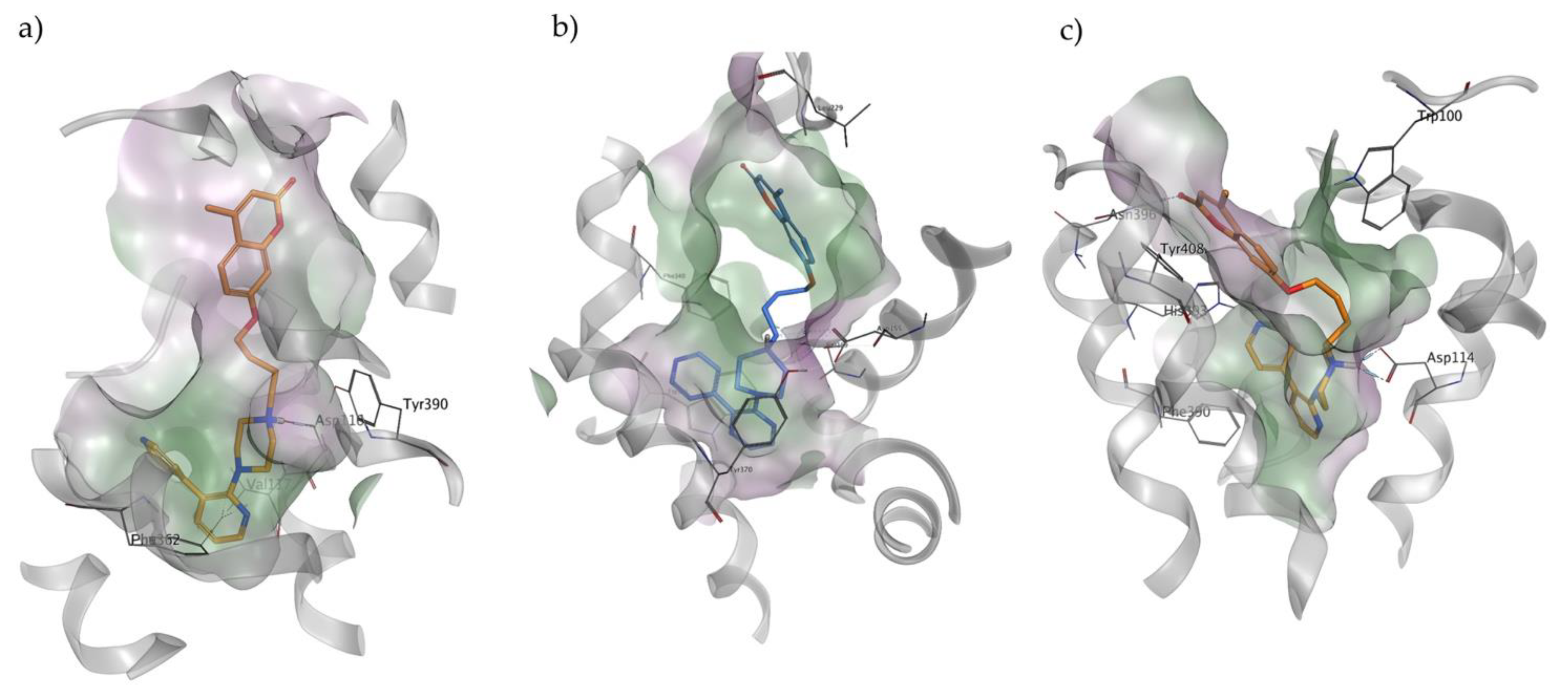
2.4. Docking Studies
2.5. In Vitro Metabolic Stability
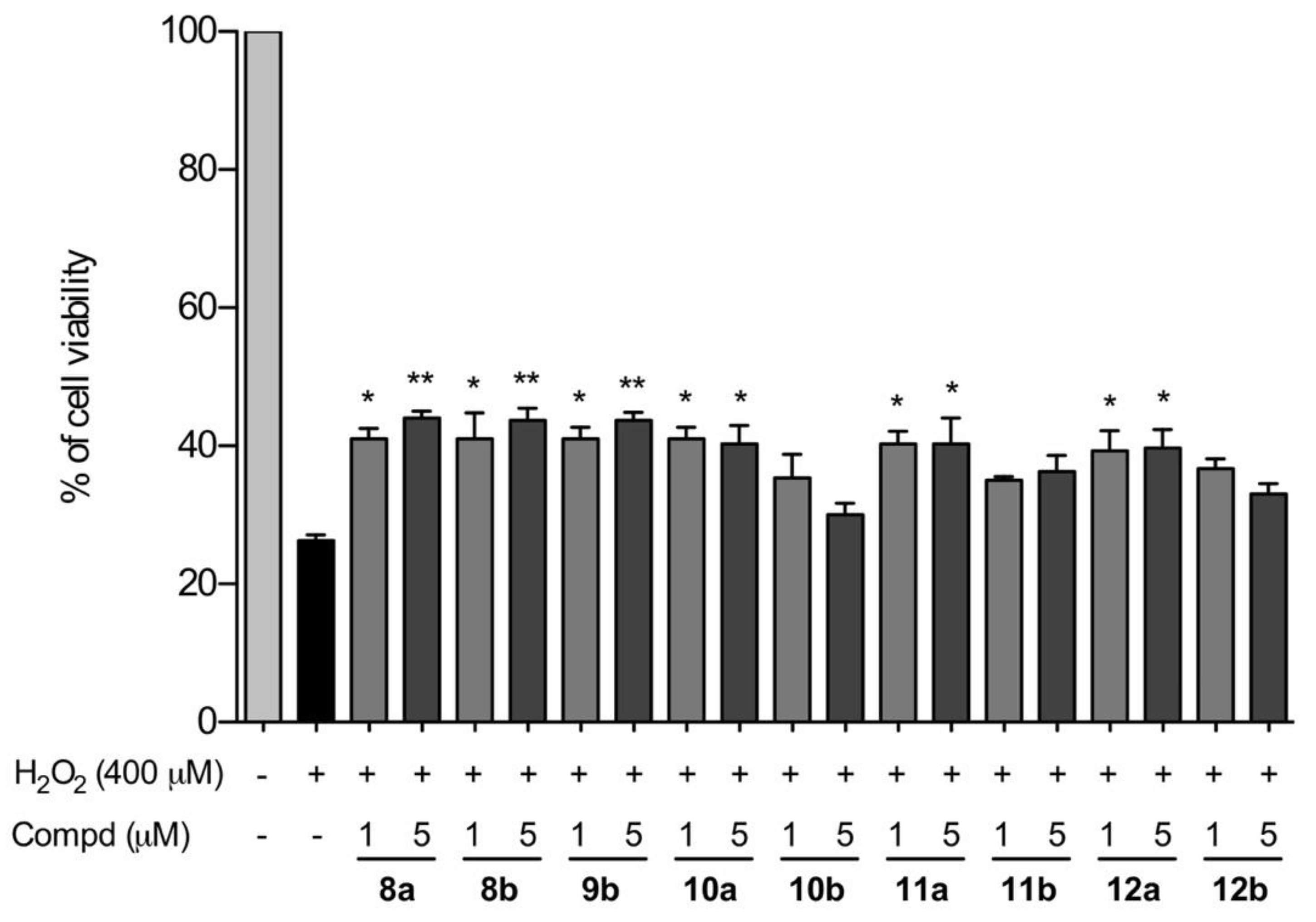
2.6. Neuroprotection against H2O2 in SHSY-5Y Cell Line
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Synthesis
4.3. Radioligand Binding Assays
4.3.1. 5-HT1A Receptor
4.3.2. 5-HT2A Receptor
4.3.3. 5-HT7 Receptor
4.3.4. Dopamine Receptors
4.4. Docking Studies
4.5. Stability Assays in Rat Liver Microsomes
4.6. Evaluation of Cell Viability
4.6.1. Cell Culture
4.6.2. Cell Viability
4.6.3. Evaluation of Cell Viability
4.6.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Meltzer, H.Y.; Massey, B.W. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 2011, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, J.A.; Mandos, L.A.; Rickels, K.; Lohoff, F.W. Pharmacological treatment of generalized anxiety disorder. Expert Opin. Pharmacother. 2011, 12, 2457–2467. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rascol, O. Piribedil for the Treatment of Motor and Non-motor Symptoms of Parkinson Disease. CNS Drugs 2016, 30, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R.U.S. FDA Approved Drugs from 2015-June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef]
- Lacivita, E.; Niso, M.; Mastromarino, M.; Garcia Silva, A.; Resch, C.; Zeug, A.; Loza, M.I.; Castro, M.; Ponimaskin, E.; Leopoldo, M. Knowledge-Based Design of Long-Chain Arylpiperazine Derivatives Targeting Multiple Serotonin Receptors as Potential Candidates for Treatment of Autism Spectrum Disorder. ACS Chem. Neurosci. 2021, 12, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Murakami, S.; Takeshima, M.; Torigoe, N.; Kitamura, Y.; Miyoshi, K. Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol. Dis. 2013, 59, 244–256. [Google Scholar] [CrossRef]
- Yuksel, T.N.; Yayla, M.; Halici, Z.; Cadirci, E.; Polat, B.; Kose, D. Protective effect of 5-HT7 receptor activation against glutamate-induced neurotoxicity in human neuroblastoma SH-SY5Y cells via antioxidative and antiapoptotic pathways. Neurotoxicol. Teratol. 2019, 72, 22–28. [Google Scholar] [CrossRef]
- Kaddouri, Y.; Abrigach, F.; Yousfi, E.B.; El Kodadi, M.; Touzani, R. New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: Synthesis, DFT calculations and molecular docking study. Heliyon 2020, 6, e03185. [Google Scholar] [CrossRef] [Green Version]
- Kucukkilinc, T.T.; Yanghagh, K.S.; Ayazgok, B.; Roknipour, M.A.; Moghadam, F.H.; Moradi, A.; Emami, S.; Amini, M.; Irannejad, H. Synthesis and neuroprotective activity of novel 1,2,4-triazine derivatives with ethyl acetate moiety against H2O2 and Aβ-induced neurotoxicity. Med. Chem. Res. 2017, 26, 3057–3071. [Google Scholar] [CrossRef]
- Largeron, M.; Mesples, B.; Gressens, P.; Cecchelli, R.; Spedding, M.; Le Ridant, A.; Fleury, M. The neuroprotective activity of 8-alkylamino-1,4-benzoxazine antioxidants. Eur. J. Pharmacol. 2001, 424, 189–194. [Google Scholar] [CrossRef]
- Robertson, A.L.; Ogryzko, N.V.; Henry, K.M.; Loynes, C.A.; Foulkes, M.J.; Meloni, M.M.; Wang, X.; Ford, C.; Jackson, M.; Ingham, P.W.; et al. Identification of benzopyrone as a common structural feature in compounds with anti-inflammatory activity in a zebrafish phenotypic screen. Dis. Model Mech. 2016, 9, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, L.; Massey, B.W.; Michael, E.; Meltzer, H.Y. Serotonin (5-HT)1A receptor agonism and 5-HT7 receptor antagonism ameliorate the subchronic phencyclidine-induced deficit in executive functioning in mice. Psychopharmacology 2016, 233, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Mayol-Llinàs, J.; Nelson, A.; Farnaby, W.; Ayscough, A. Assessing molecular scaffolds for CNS drug discovery. Drug Discov. Today 2017, 22, 965–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolyasnikova, K.N.; Vichuzhanin, M.V.; Konstantinopol’skii, M.A.; Trofimov, S.S.; Gudasheva, T.A. Synthesis and pharmacological activity of analogs of the endogenous neuropeptide cycloprolylglycine. Pharma. Chem. J. 2012, 46, 96–102. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [PubMed]
- Lacivita, E.; Podlewska, S.; Speranza, L.; Niso, M.; Satała, G.; Perrone, R.; Perrone-Capano, C.; Bojarski, A.J.; Leopoldo, M. Structural modifications of the serotonin 5-HT7 receptor agonist N-(4-cyanophenylmethyl)-4-(2-biphenyl)-1-piperazinehexanamide (LP-211) to improve in vitro microsomal stability: A case study. Eur. J. Med. Chem. 2016, 120, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.W.; Gangloff, A.R.; Jennings, A.J.; Vu, P.H. Poly (ADP-Ribose) Polymerase (PARP) Inhibitors. International Patent Application No. PCT/US2010/028878, 30 September 2010. [Google Scholar]
- Kumar, J.S.; Majo, V.J.; Hsiung, S.C.; Millak, M.S.; Liu, K.P.; Tamir, H.; Prabhakaran, J.; Simpson, N.R.; Van Heertum, R.L.; Mann, J.J.; et al. Synthesis and in vivo validation of [O-methyl-11C]2-{4-[4-(7-methoxynaphthalen-1-yl)piperazin- 1-yl]butyl}-4-methyl-2H-[1,2,4]triazine-3,5-dione: A novel 5-HT1A receptor agonist positron emission tomography ligand. J. Med. Chem. 2006, 49, 125–134. [Google Scholar] [CrossRef]
- Hansen, H.D.; Lacivita, E.; Di Pilato, P.; Herth, M.M.; Lehel, S.; Ettrup, A.; Andersen, V.L.; Dyssegaard, A.; De Giorgio, P.; Perrone, R.; et al. Synthesis, radiolabeling and in vivo evaluation of [(11)C](R)-1-[4-[2-(4-methoxyphenyl)phenyl]piperazin-1-yl]-3-(2-pyrazinyloxy)-2-propanol, a potential PET radioligand for the 5-HT(7) receptor. Eur. J. Med. Chem. 2014, 79, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Silva, A.G.; Loza, M.I.; Kolb, P.; Castro, M.; Poso, A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. ChemMedChem 2016, 11, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Varin, T.; Gutiérrez-de-Terán, H.; Castro, M.; Brea, J.; Fabis, F.; Dauphin, F.; Aqvist, J.; Lepailleur, A.; Perez, P.; Burgueño, J.; et al. Phe369(7.38) at human 5-HT(7) receptors confers interspecies selectivity to antagonists and partial agonists. Br. J. Pharmacol. 2010, 159, 1069–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.; Kiss, D.J.; Keserű, G.M.; Stark, H. Binding kinetics of cariprazine and aripiprazole at the dopamine D3 receptor. Sci. Rep. 2018, 8, 12509. [Google Scholar] [CrossRef]
- Bautista-Aguilera, Ó.M.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3R Antagonism for Neurodegenerative Diseases. Angew. Chem. Int. Ed. 2017, 56, 12765. [Google Scholar] [CrossRef] [Green Version]
- Schübler, M.; Sadek, B.; Kottke, T.; Weizel, L.; Stark, H. Synthesis, Molecular Properties Estimations, and Dual Dopamine D2 and D3 Receptor Activities of Benzothiazole-Based Ligands. Front. Chem. 2017, 5, 64. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.D.; Yen, H.Y.; Robinson, C.V.; et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021, 592, 469–473. [Google Scholar] [CrossRef]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell 2020, 182, 1574–1588.e19. [Google Scholar] [CrossRef]
- Sun, B.; Feng, D.; Chu, M.L.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Pati, M.L.; Hornick, J.R.; Niso, M.; Berardi, F.; Spitzer, D.; Abate, C.; Hawkins, W. Sigma-2 receptor agonist derivatives of 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) induce cell death via mitochondrial superoxide production and caspase activation in pancreatic cancer. BMC Cancer 2017, 17, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, N.H.; Phopin, K.; Suwanjang, W.; Songtawee, N.; Ruankham, W.; Wongchitrat, P.; Prachayasittikul, S.; Prachayasittikul, V. Neuroprotective Effects of Phenolic and Carboxylic Acids on Oxidative Stress-Induced Toxicity in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2018, 43, 619–636. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Structure | cLogP 1 | MPO 2 | MS 3 | |
|---|---|---|---|---|---|
| 8a | n = 3 |  | 2.79 | 4.98 | 4% |
| 8b | n = 4 | 3.31 | 4.54 | 14% | |
| 9a | n = 3 |  | 3.06 | 5.05 | <3% |
| 9b | n = 4 | 3.58 | 4.47 | 45% | |
| 10a | n = 3 |  | 3.08 | 5.07 | 5% |
| 10b | n = 4 | 3.60 | 4.49 | 26% | |
| 11a | n = 3 |  | 1.24 | 5.66 | 4% |
| 11b | n = 4 | 1.76 | 5.44 | 3% | |
| 12a | n = 3 |  | 3.55 | 4.52 | 19% |
| 12b | n = 4 | 4.07 | 3.97 | 14% |
| Compd | Affinity | Cytotoxicity | ||||||
|---|---|---|---|---|---|---|---|---|
| Ki [nM] ± S.E.M. | Ki [nM] (CI 95%) | EC50 [μM] ± S.E.M. | ||||||
| 5-HT1A 1 | 5-HT2A 1 | 5-HT7 1 | D2 | D3 | D1 | D5 | ||
| 8a | 13.1 ± 2.3 | 782 ± 119 | 10.7 ± 0.7 | 388 (249; 606) | 1223 (684; 2185) | 6290 (1608; 24,595) | 7412 (2038; 26,956) | 32.9 ± 3.5 |
| 8b | 17.6 ± 0.9 | 611 ± 87 | 9.38 ± 0.42 | 9.08 (5.07; 16.2) | 691 (516; 926) | 1711 (981; 2983) | 8205 (2475; 27,203) | 25.5 ± 2.2 |
| 9a | 653 ± 95 | 49.1 ± 12 | 60.8 ± 7.1 | >10,000 | >10,000 | 193 (137; 271) | 413 (199; 856) | nd |
| 9b | 23.9 ± 4.9 | 39.4 ± 7.4 | 45.0 ± 3.4 | >10,000 | >10,000 | 88.3 (35.7; 218) | 222 (76.0; 646) | 51.9 ± 4.3 |
| 10a | 1091 ± 154 | 96.8 ± 11 | 45.7 ± 1.3 | >10,000 | >10,000 | 104 (33.0; 331) | 180 (96.2; 338) | 28.2 ± 3.2 |
| 10b | 43.4 ± 6.2 | 80.7 ± 3.1 | 36.5 ± 5.2 | >10,000 | >10,000 | 140 (73.4; 268) | 101 (33.8; 299) | 34.3 ± 2.5 |
| 11a | 269 ± 18 | >10,000 | 44.5 ± 2.2 | >10,000 | >10,000 | >10,000 | >10,000 | >100 |
| 11b | 5.19 ± 0.12 | >10,000 | 79.4 ± 9.0 | >10,000 | >10,000 | >10,000 | >10,000 | >100 |
| 12a | 41.5 ± 3.0 | 315 ± 44 | 42.5 ± 6.2 | 300 (210; 430) | 1075 (651; 1776) | 2198 (780; 6191) | >10,000 | 35.2 ± 4.1 |
| 12b | 11.3 ± 0.4 | ~1419 2 | 52.2 ± 10.6 | 17.0 (4.56; 63.5) | 424 (201; 960) | 1527 (354; 6586) | 2733 (683; 10,929) | 47.9 ± 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastromarino, M.; Niso, M.; Abate, C.; Proschak, E.; Dubiel, M.; Stark, H.; Castro, M.; Lacivita, E.; Leopoldo, M. Design and Synthesis of Arylpiperazine Serotonergic/Dopaminergic Ligands with Neuroprotective Properties. Molecules 2022, 27, 1297. https://doi.org/10.3390/molecules27041297
Mastromarino M, Niso M, Abate C, Proschak E, Dubiel M, Stark H, Castro M, Lacivita E, Leopoldo M. Design and Synthesis of Arylpiperazine Serotonergic/Dopaminergic Ligands with Neuroprotective Properties. Molecules. 2022; 27(4):1297. https://doi.org/10.3390/molecules27041297
Chicago/Turabian StyleMastromarino, Margherita, Mauro Niso, Carmen Abate, Ewgenij Proschak, Mariam Dubiel, Holger Stark, Marián Castro, Enza Lacivita, and Marcello Leopoldo. 2022. "Design and Synthesis of Arylpiperazine Serotonergic/Dopaminergic Ligands with Neuroprotective Properties" Molecules 27, no. 4: 1297. https://doi.org/10.3390/molecules27041297