
Diels–Alder Cycloaddition Reactions in Sustainable Media




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bio-Based Solvents
2.1. Glycerol
2.2. Gluconic Acid
3. Polyethylene Glycol
4. Organic Carbonates
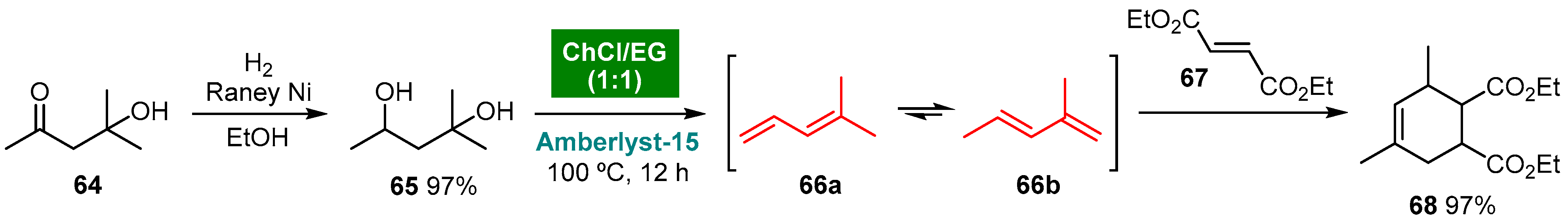
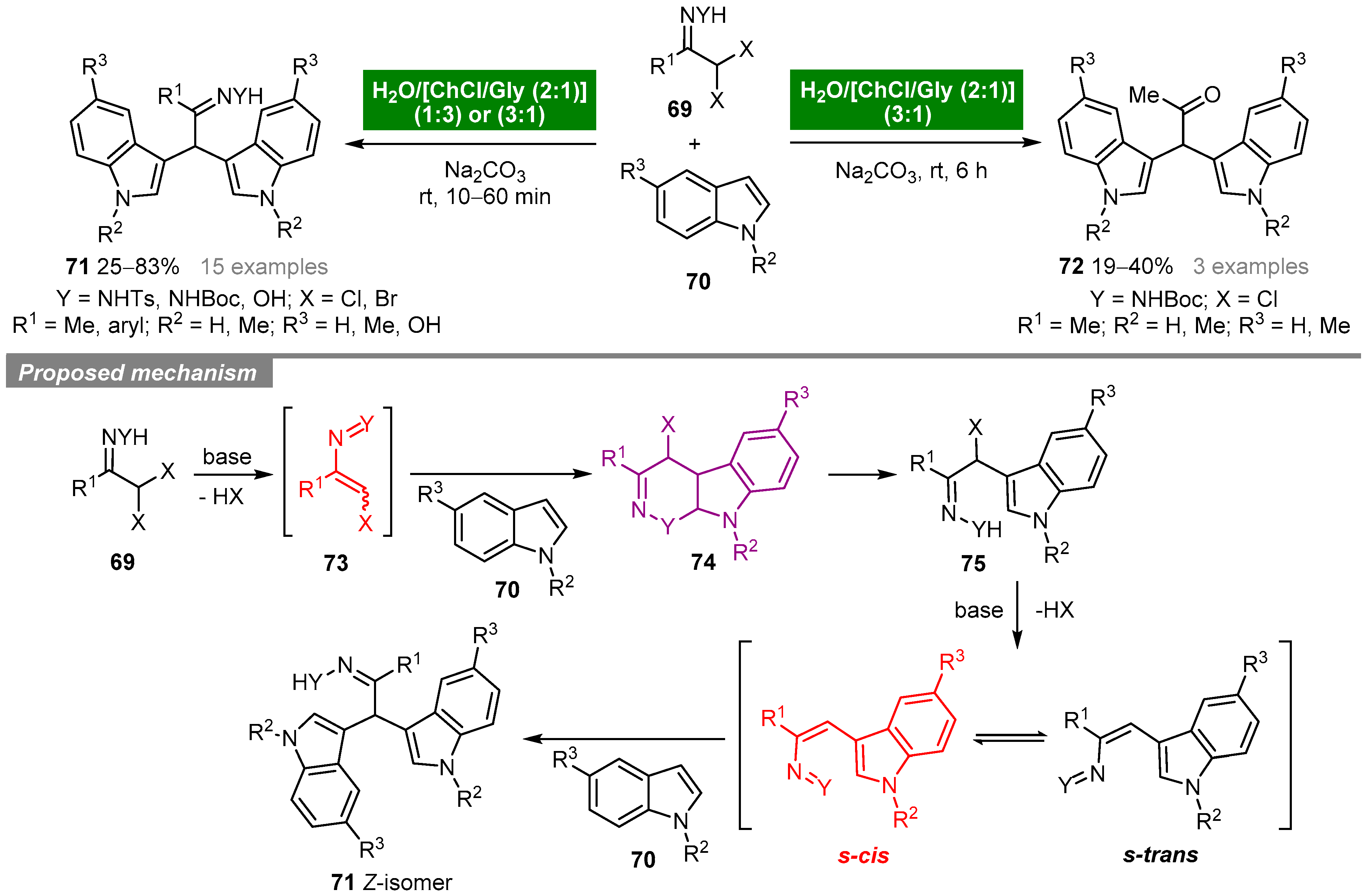
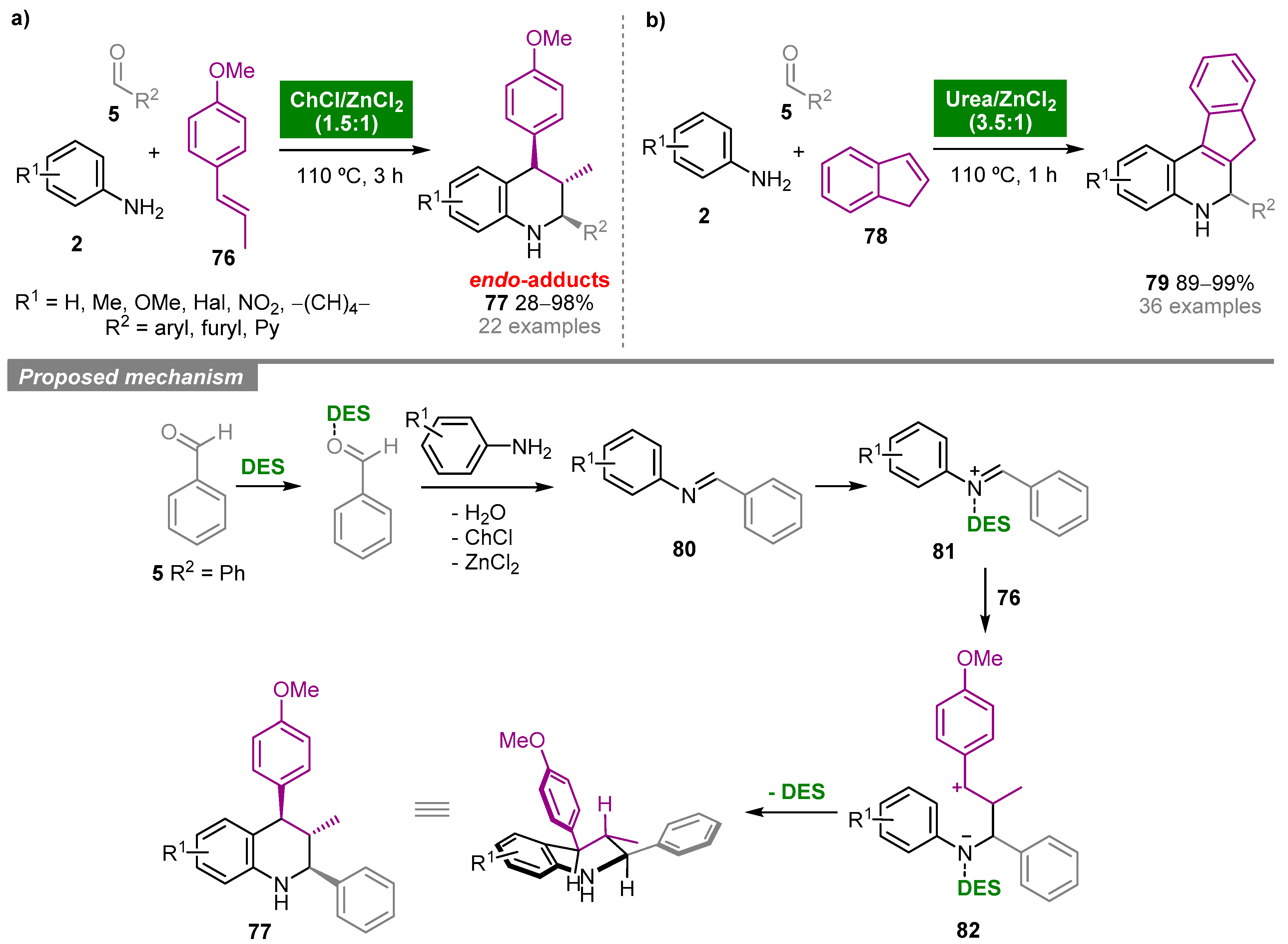
5. Deep Eutectic Solvents
6. Supercritical Carbon Dioxide
7. Water
7.1. The Water Effect: Experimental and Theoretical Studies
7.2. Noncatalyzed Diels–Alder Cycloaddition Reactions
7.3. Catalyzed Diels–Alder Cycloaddition Reactions
7.4. Asymmetric Diels–Alder Cycloaddition Reactions
7.5. Total Synthesis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis, 1st ed.; Pergamon Press: Oxford, UK, 1990; Volume 8, p. 373. [Google Scholar]
- Hayashi, Y. Catalytic asymmetric Diels–Alder reactions. In Cycloaddition Reactions in Organic Synthesis; Kobayashi, K.A.J.S., Ed.; Wiley-VCH: Weinheim, Germany, 2002; pp. 5–55. [Google Scholar]
- Pindur, U.; Lutz, G.; Otto, C. Acceleration and selectivity enhancement of Diels–Alder reactions by special and catalytic methods. Chem. Rev. 1993, 93, 741–761. [Google Scholar] [CrossRef]
- Corey, E.J. Catalytic enantioselective Diels–Alder reactions: Methods, mechanistic fundamentals, pathways, and applications. Angew. Chem. Int. Ed. Engl. 2002, 41, 1650–1667. [Google Scholar] [CrossRef]
- Merino, P.; Marqués-López, E.; Tejero, T.; Herrera, R.P. Enantioselective organocatalytic Diels–Alder reactions. Synthesis 2010, 1, 1–26. [Google Scholar] [CrossRef]
- Laína-Martin, V.; Fernández-Salas, J.A.; Alemán, J. Organocatalytic strategies for the development of the enantioselective inverse-electron-demand hetero-Diels–Alder reaction. Chem. Eur. J. 2021, 27, 12509–12520. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy—A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Lam, C.H.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.S.; et al. The Green ChemisTREE: 20 Years after taking root with the 12 principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green solvents for sustainable organic synthesis: State of the art. Green Chem. 2005, 7, 267–278. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C. Greener media in chemical synthesis and processing. Angew. Chem. Int. Ed. 2006, 45, 3904–3908. [Google Scholar] [CrossRef]
- Andrade, C.K.Z.; Alves, L.M. Environmentally benign solvents in organic synthesis: Current topics. Curr. Org. Chem. 2005, 9, 195–218. [Google Scholar] [CrossRef]
- Welton, T. Solvents and sustainable chemistry. Proc. R. Soc. A 2015, 471, 20150502. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and sustainable solvents in chemical processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Wagare, D.S.; Shirsath, S.E.; Shaikh, M.; Netankar, P. Sustainable solvents in chemical synthesis: A review. Environ. Chem. Lett. 2021, 19, 3263–3282. [Google Scholar] [CrossRef]
- Tanaka, K.; Toda, F. Solvent-free organic synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.L.; Jérôme, F. Bio-based solvents: An emerging generation of fluids for the design of eco-efficient processes in catalysis and organic chemistry. Chem. Soc. Rev. 2013, 42, 9550–9570. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Ferlin, F.; Luciani, L.; Ackermann, L.; Vaccaro, L. Biomass-derived solvents as effective media for cross-coupling reactions and C−H functionalization processes. Green Chem. 2017, 19, 1601–1612. [Google Scholar] [CrossRef]
- Sydnes, M.O. Green bio-based solvents in C–C cross-coupling reactions. Curr. Green Chem. 2019, 6, 96–104. [Google Scholar] [CrossRef]
- Gandeepan, P.; Kaplaneris, N.; Santoro, S.; Vaccaro, L.; Ackermann, L. Biomass-derived solvents for sustainable transition metal-catalyzed C–H activation. ACS Sustain. Chem. Eng. 2019, 7, 8023–8040. [Google Scholar] [CrossRef]
- Chen, J.; Spear, S.K.; Huddleston, J.G.; Rogers, R.D. Polyethylene glycol and solutions of polyethylene glycol as green reaction media. Green Chem. 2005, 7, 64–82. [Google Scholar]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef]
- Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Zanatta, N.; Bonacorso, H.G. Ionic liquids in heterocyclic synthesis. Chem. Rev. 2008, 108, 2015–2050. [Google Scholar] [CrossRef]
- Wang, A.; Zheng, X.; Zhao, Z.; Li, C.; Zheng, X. Deep eutectic solvents to organic synthesis. Prog. Chem. 2014, 26, 784–795. [Google Scholar]
- Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Khandelwal, S.; Tailor, Y.K.; Kumar, M. Deep eutectic solvents (DESs) as eco-friendly and sustainable solvent/catalyst systems in organic transformations. J. Mol. Liq. 2016, 215, 345–386. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Deep eutectic solvents: The organic reaction medium of the century. Eur. J. Org. Chem. 2016, 2016, 612–632. [Google Scholar]
- Kaupp, G. Reactions in supercritical carbon dioxide. Angew. Chem. Int. Ed. 1994, 33, 1452–1455. [Google Scholar] [CrossRef]
- Oakes, R.S.; Clifford, A.A.; Rayner, C.M. The use of supercritical fluids in synthetic organic chemistry. J. Chem. Soc. Perkin Trans. 2001, 1, 917–941. [Google Scholar] [CrossRef]
- Licence, P.; Ke, J.; Sokolova, M.; Ross, S.K.; Poliakoff, M. Chemical reactions in supercritical carbon dioxide: From laboratory to commercial plant. Green Chem. 2003, 5, 99–104. [Google Scholar] [CrossRef]
- Beckman, E.J. Supercritical and near-critical CO2 in green chemical synthesis and processing. J. Supercrit. Fluids 2004, 28, 121–191. [Google Scholar] [CrossRef]
- Savage, P.E.; Gopalan, S.; Mizan, T.I.; Martino, C.J.; Brock, E.E. Reactions at supercritical conditions: Applications and fundamentals. AIChE J. 1995, 41, 1723–1778. [Google Scholar] [CrossRef] [Green Version]
- Hailes, H.C. Reaction solvent selection: The potential of water as a solvent for organic transformations. Org. Process Res. Dev. 2007, 11, 114–120. [Google Scholar] [CrossRef]
- Chanda, A.; Fokin, V.V. Organic synthesis “on water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef] [Green Version]
- Gawande, M.B.; Bonifácio, V.D.B.; Luque, R.; Branco, P.S.; Varma, R.S. Benign by design: Catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 2013, 42, 5522–5551. [Google Scholar]
- Butler, R.N.; Coyne, A.G. Organic synthesis reactions on-water at the organic-liquid water interface. Org. Biomol. Chem. 2016, 14, 9945–9960. [Google Scholar] [CrossRef] [Green Version]
- Kitanosono, T.; Masuda, K.; Xu, P.Y.; Kobayashi, S. Catalytic organic reactions in water toward sustainable society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef]
- Bhowmick, K.C.; Bihani, M.; Zhao, J.C.-G. Organocatalyzed asymmetric Diels–Alder reactions in aqueous or semi-aqueous media. Mini Rev. Org. Chem. 2018, 15, 3–19. [Google Scholar] [CrossRef]
- Brummond, K.M.; Wach, C.K. Environmentally benign solvent systems: Toward a greener [4+2] cycloaddition process. Mini-Rev. Org. Chem. 2007, 4, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Aridoss, G.; Laali, K.K. Ionic liquids as novel media and catalysts for Diels–Alder chemistry. Curr. Org. Synth. 2017, 14, 952–971. [Google Scholar]
- Gupta, G.R.; Girase, T.R.; Kapdi, A.R. Ionic liquid as a sustainable reaction medium for Diels–Alder reaction. In Encyclopedia of Ionic Liquids; Zhang, S., Ed.; Springer: Singapore, 2019. [Google Scholar]
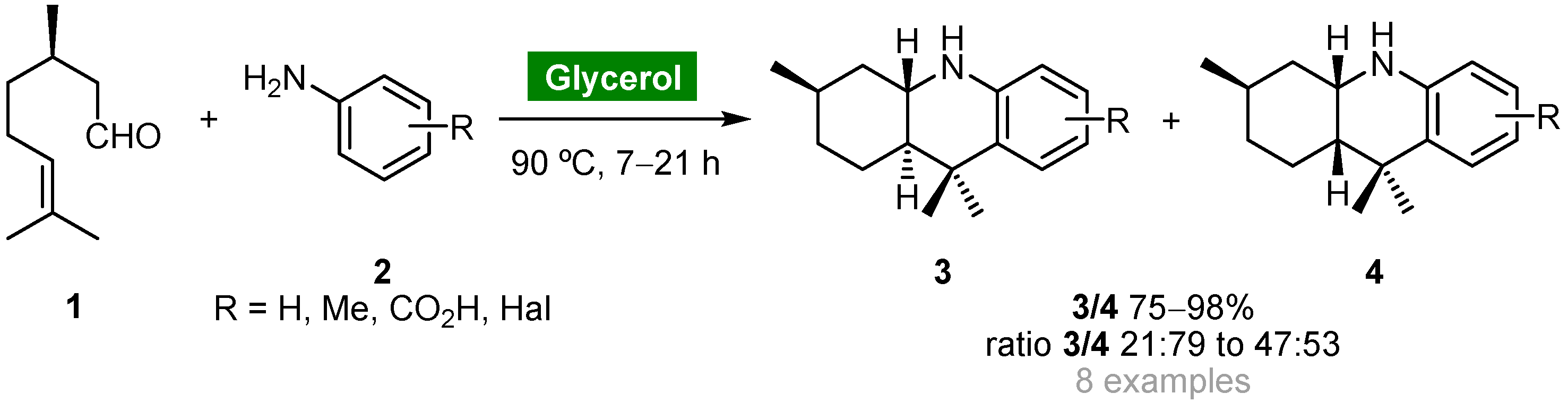
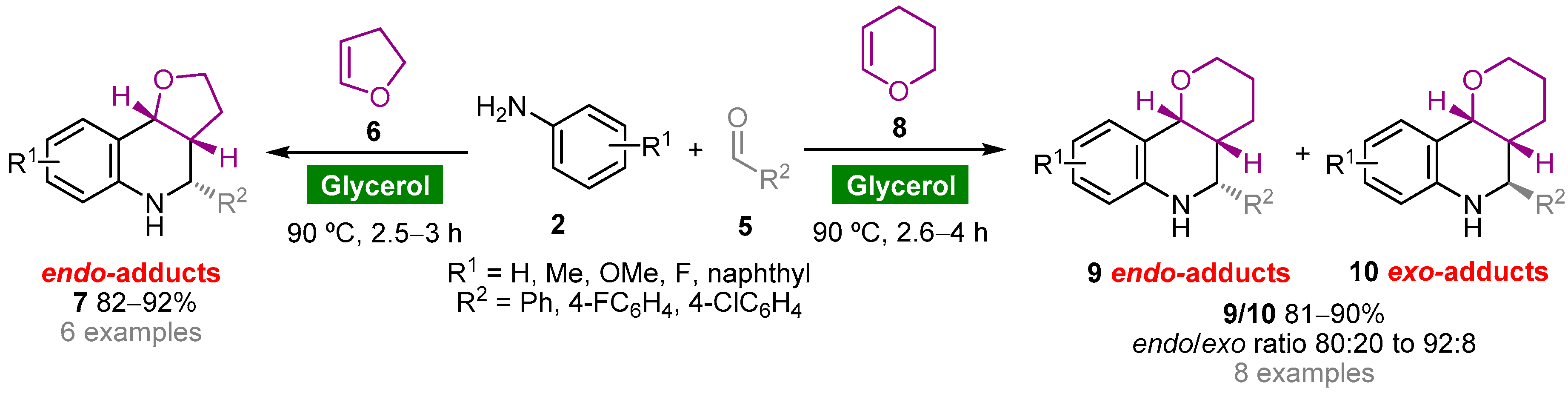
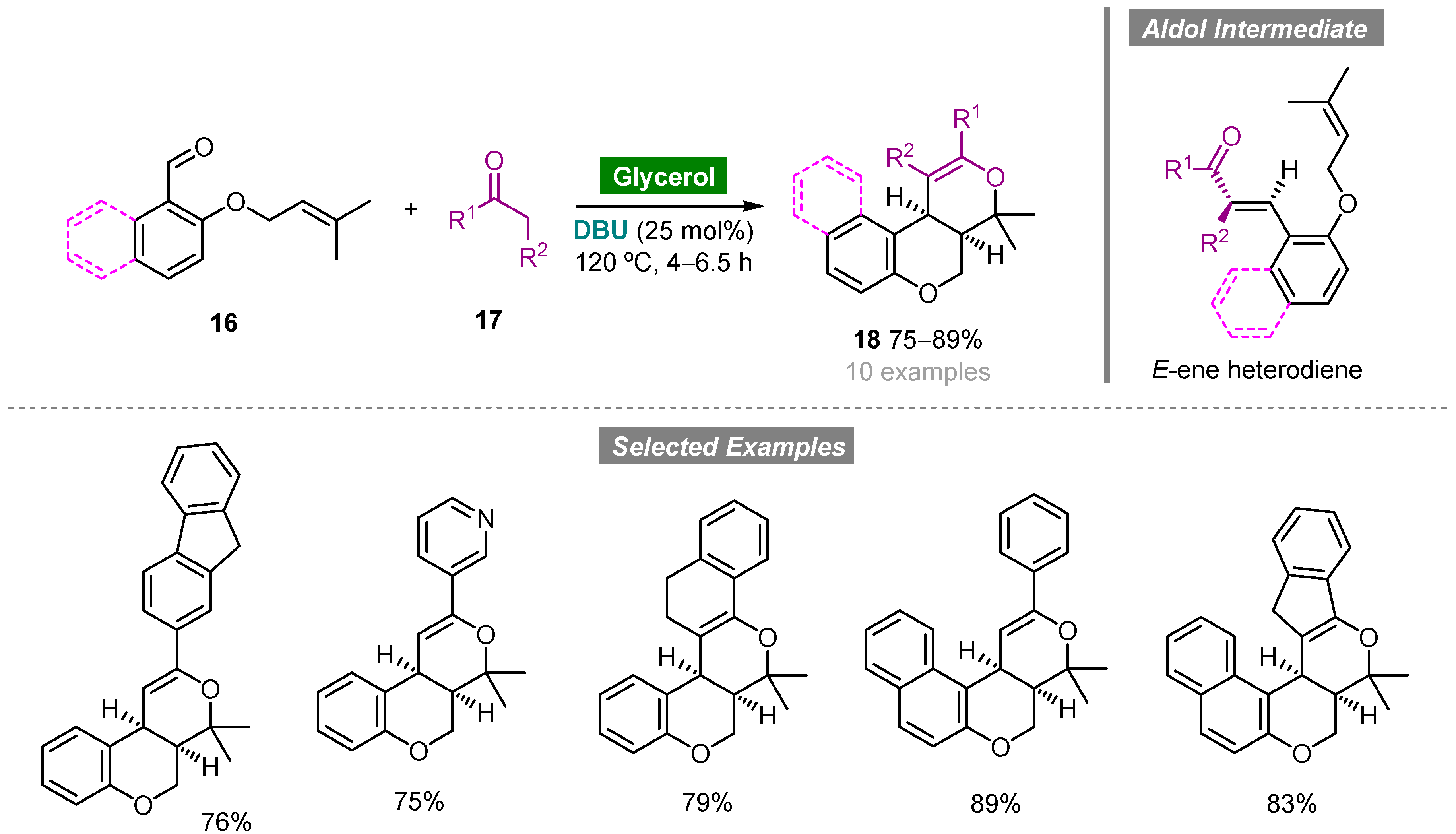
- Díaz-Álvarez, A.E.; Francos, J.; Lastra-Barreira, B.; Crochet, P.; Cadierno, V. Glycerol and derived solvents: New sustainable reaction media for organic synthesis. Chem. Commun. 2011, 47, 6208–6227. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Álvarez, A.E.; Francos, J.; Crochet, P.; Cadierno, V. Recent advances in the use of glycerol as green solvent for synthetic organic chemistry. Curr. Green Chem. 2014, 1, 51–65. [Google Scholar] [CrossRef]
- Abd-Elmonem, M.; Mekheimer, R.A.; Hayallah, A.M.; Abo-Elsoud, F.; Sadek, K.U. Recent advances in the utility of glycerol as a benign and biodegradable medium in heterocyclic synthesis. Curr. Org. Chem. 2019, 23, 3226–3246. [Google Scholar] [CrossRef]
- Kouznetsov, V.V. Recent synthetic developments in a powerful imino Diels–Alder reaction (Povarov reaction): Application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron 2009, 65, 2721–2750. [Google Scholar] [CrossRef]
- Fochi, M.; Caruana, L.; Bernardi, L. Catalytic asymmetric aza-Diels–Alder reactions: The Povarov cycloaddition reaction. Synthesis 2014, 46, 135–157. [Google Scholar] [CrossRef]
- Nascimento, J.E.R.; Barcellos, A.M.; Sachini, M.; Perin, G.; Lenardão, E.J.; Alves, D.; Jacob, R.G.; Missau, F. Catalyst-free synthesis of octahydroacridines using glycerol as recyclable solvent. Tetrahedron Lett. 2011, 52, 2571–2574. [Google Scholar] [CrossRef] [Green Version]
- Somwanshi, J.L.; Shinde, N.D.; Faruqui, M. Catalyst-free synthesis of furano- and pyranoquinolines by using glycerol as recyclabe solvent. Heterocycl. Lett. 2013, 3, 69–74. [Google Scholar]
- Majumdar, K.C.; Taher, A.; Nandi, R.K. Synthesis of heterocycles by domino Knoevenagel−hetero-Diels–Alder reactions. Tetrahedron 2012, 68, 5693–5718. [Google Scholar] [CrossRef]
- Parmar, N.J.; Barad, H.A.; Labana, B.M.; Kant, R.; Gupta, V.K. A glycerol mediated domino reaction: An efficient, green synthesis of polyheterocycles incorporating a new thiochromeno[2,3-b]quinoline unit. RSC Adv. 2013, 3, 20719–20725. [Google Scholar] [CrossRef]
- Parmar, B.D.; Sutariya, T.R.; Brahmbhatt, G.C.; Parmar, N.J.; Kant, R.; Gupta, V.K. A base-catalyzed, domino aldol/hetero-Diels–Alder synthesis of tricyclic pyrano[3,4-c]chromenes in glycerol. J. Org. Chem. 2016, 81, 4955–4964. [Google Scholar] [CrossRef]
- Lim, H.Y.; Dolzhenko, A.V. Gluconic acid aqueous solution: A bio-based catalytic medium for organic synthesis. Sustain. Chem. Pharm. 2021, 21, 100443. [Google Scholar] [CrossRef]
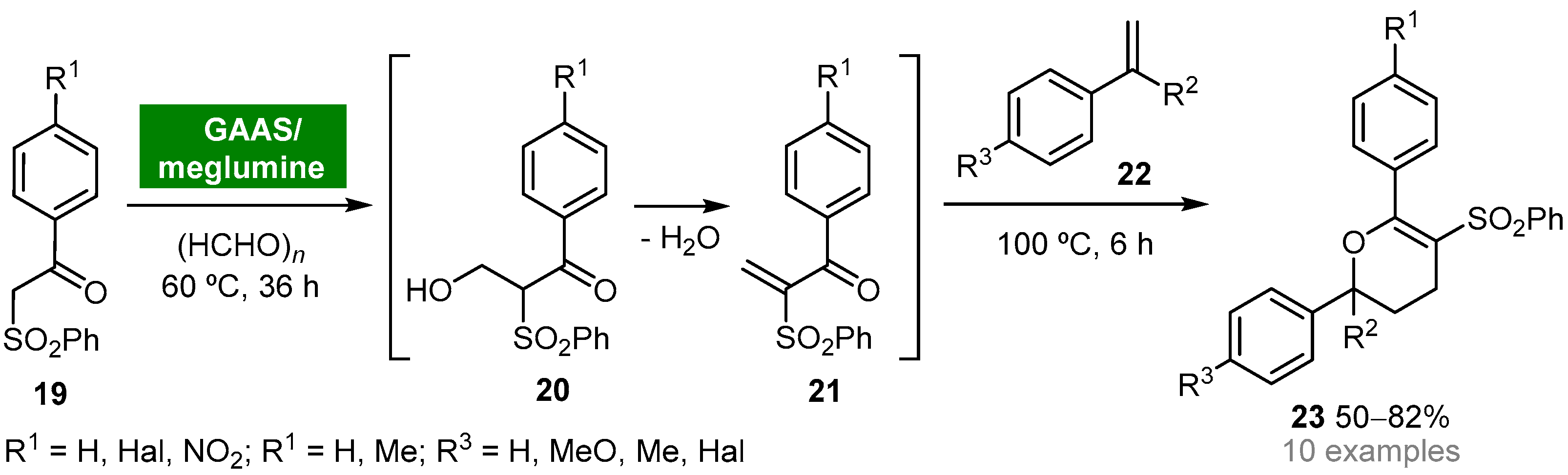
- Yang, J.; Li, H.; Li, M.; Peng, J.; Gu, Y. Multicomponent reactions of β-ketosulfones and formaldeyde in a bio-based binary mixture solvent system composed of meglumine and gluconic acid aqueous solution. Adv. Synth. Catal. 2012, 354, 688–700. [Google Scholar] [CrossRef]
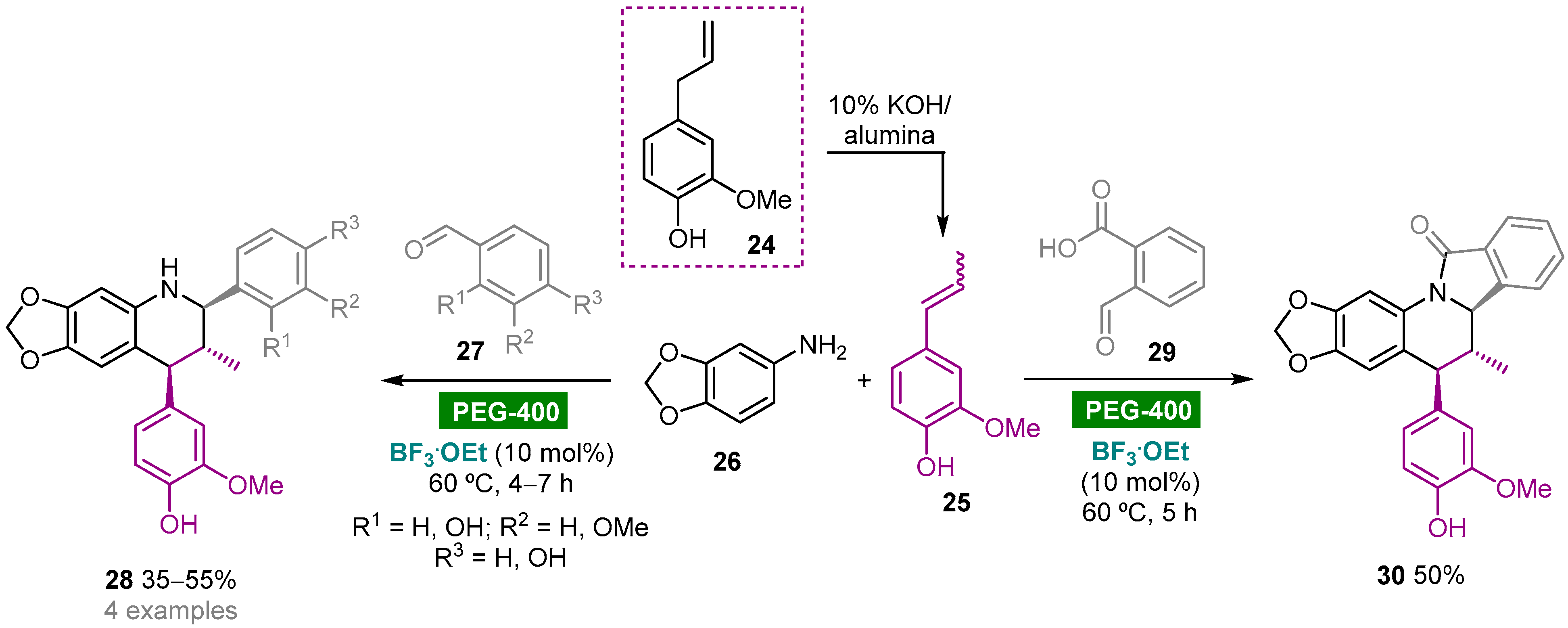
- Arenas, D.R.M.; Ruíz, F.A.R.; Kouznetsov, V.V. Highly diastereoselective synthesis of new heterolignan-like 6,7-methylendioxy-tetrahydroquinolines using the clove bud essential oil as raw material. Tetrahedron Lett. 2011, 52, 1388–1391. [Google Scholar] [CrossRef]
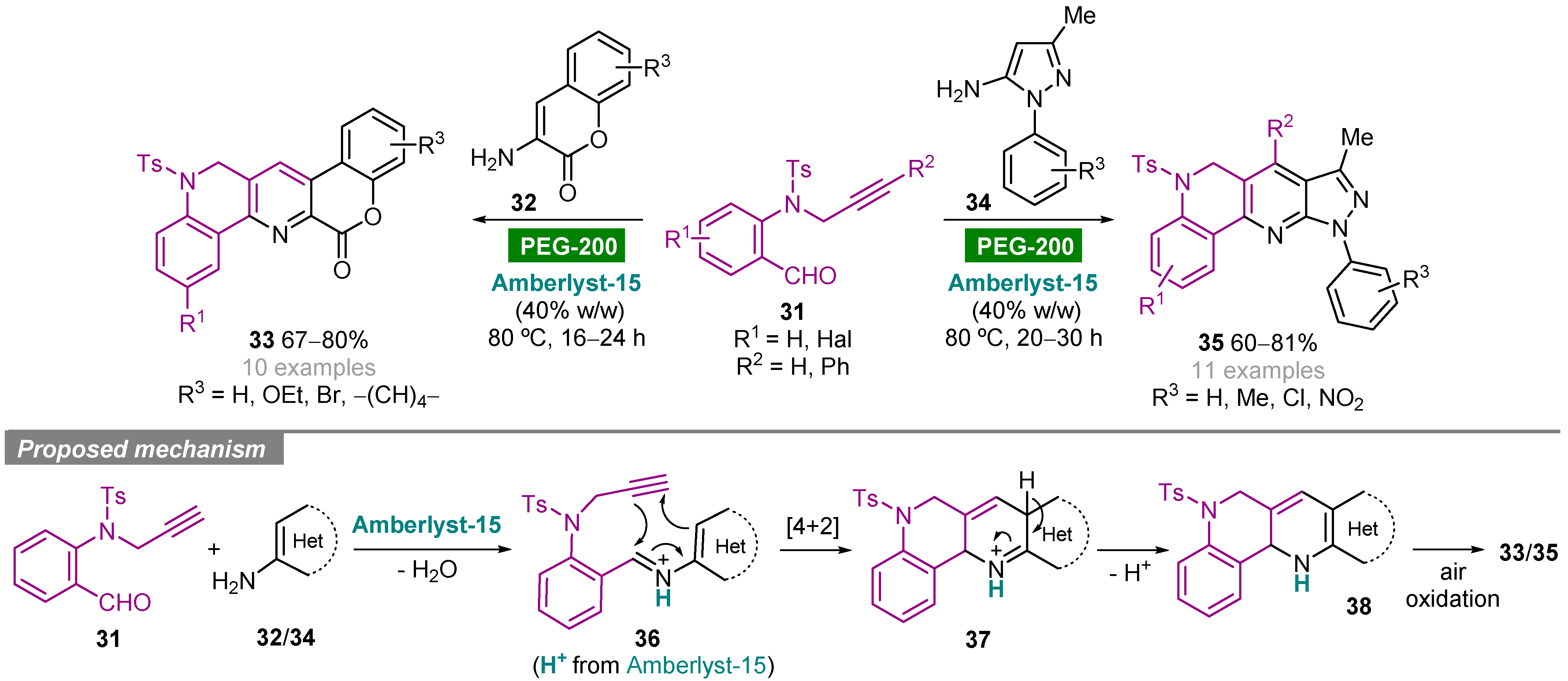
- Muthukrishnan, I.; Vachan, B.S.; Karuppasamy, M.; Eniyaval, A.; Maheswari, C.U.; Nagarajan, S.; Menéndez, J.C.; Sridharan, V. Heterogeneous Amberlyst-15-catalyzed synthesis of complex hybrid heterocycles containing 1,6-naphthyridine under metal-free green conditions. Org. Biomol. Chem. 2019, 17, 6872–6879. [Google Scholar] [CrossRef]
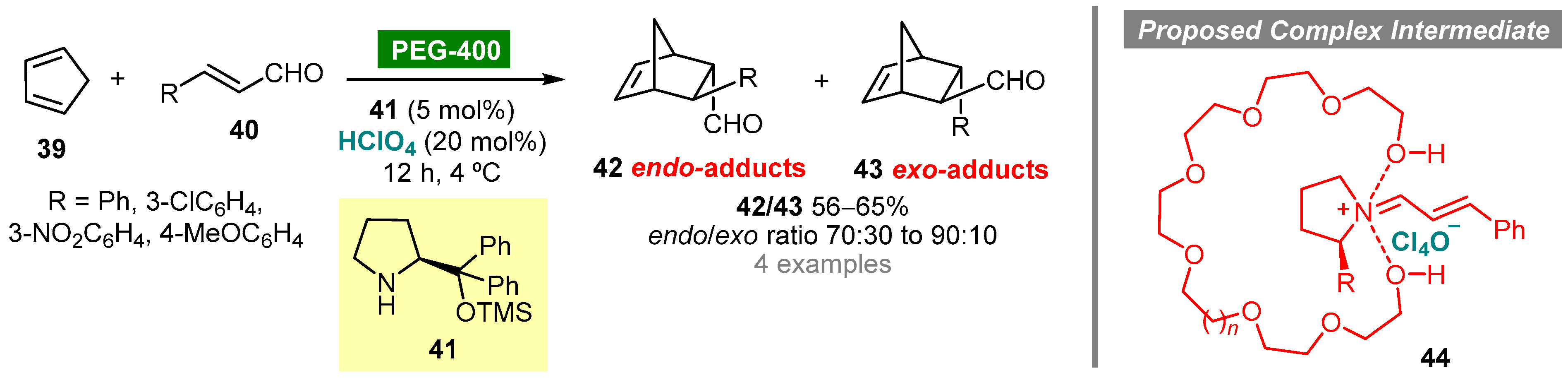
- Anitha, P.M.; Mainkar, P.S.; Kallepu, S.; Babu, V.S.P.; Reddy, C.S.; Chandrasekhar, S. Caveat in the stereochemical outcome of the organocatalytic Diels–Alder reaction in PEG-400. RSC Adv. 2016, 6, 76132–76136. [Google Scholar] [CrossRef]
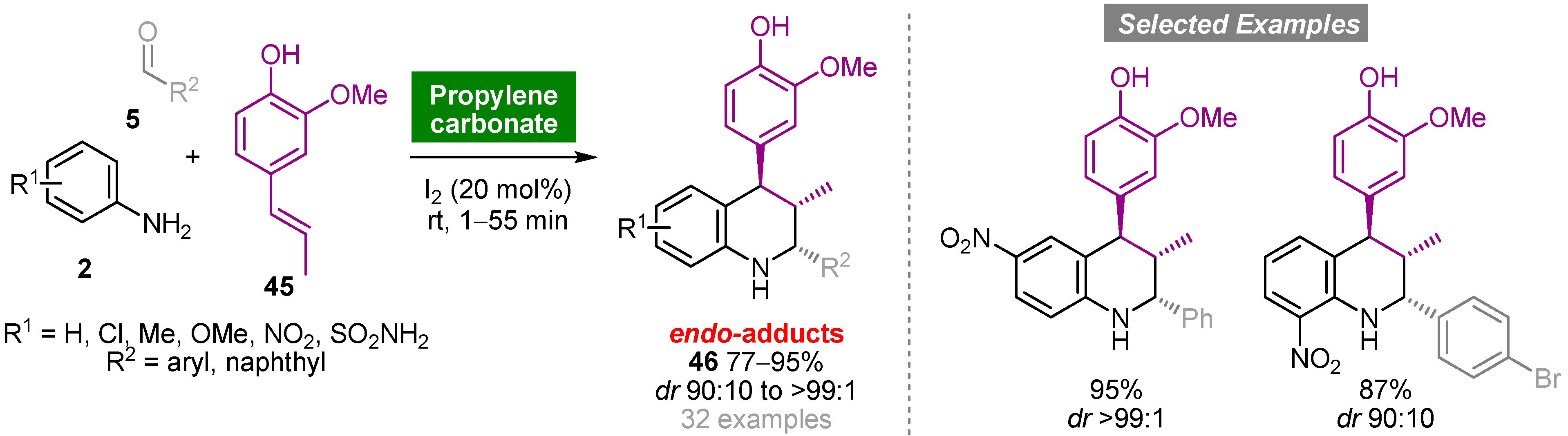
- Forero, J.S.B.; Muñoz, J.A.H.; Jones, J.; da Silva, F.M. Propylene carbonate in organic synthesis: Exploring its potential as a green solvent. Curr. Org. Synth. 2016, 13, 834–846. [Google Scholar] [CrossRef]
- Forero, J.S.B.; de Carvalho, E.M.; Junior, J.J.; da Silva, F.M. Facile, efficient diastereoselective synthesis of tetrahydroquinoline scaffolds using propylene carbonate as an eco-friendly solvent. Curr. Org. Synth. 2015, 12, 102–107. [Google Scholar] [CrossRef]
- Aricò, F.; Tundo, P. Dimethylcarbonate: A modern green reagent and solvent. Russ. Chem. Rev. 2010, 79, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, G.; Perosa, A.; Selva, M. Dimethyl carbonate: A versatile reagent for a sustainable valorization of renewables. Green Chem. 2018, 20, 288–322. [Google Scholar] [CrossRef]
- Meng, D.; Li, D.Z.; Ollevier, T. Recyclable iron(II) caffeine-derived ionic salt catalyst in the Diels–Alder reaction of cyclopentadiene and α,β-unsaturated N-acyl-oxazolidinones in dimethyl carbonate. RSC Adv. 2019, 9, 21956–21963. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A.P.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef]
- Zhang, Q.; Vigier, K.O.; Royer, S.; Jérôme, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep eutectic solvents (DESs) and their applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Xue, Z.; Mu, T. Eutectics: Formation, properties, and applications. Chem. Soc. Rev. 2021, 50, 8596–8638. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep eutectic solvents: A review of fundamentals and applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Nagare, A.S.; Kumar, A. Eutectic mixture-directed kinetics of Diels–Alder reaction. Indian J. Chem. Sect. A 2011, 50, 788–792. [Google Scholar]
- Kumar, A.; Deshpande, S.S. Experimental evidence to support viscosity dependence of rates of Diels–Alder reactions in solvent media. J. Org. Chem. 2003, 68, 5411–5414. [Google Scholar] [CrossRef] [PubMed]
- Marullo, S.; Meli, A.; D’Anna, F. A joint action of deep eutectic solvents and ultrasound to promote Diels–Alder reaction in a sustainable way. ACS Sustain. Chem. Eng. 2020, 8, 4889–4899. [Google Scholar] [CrossRef]
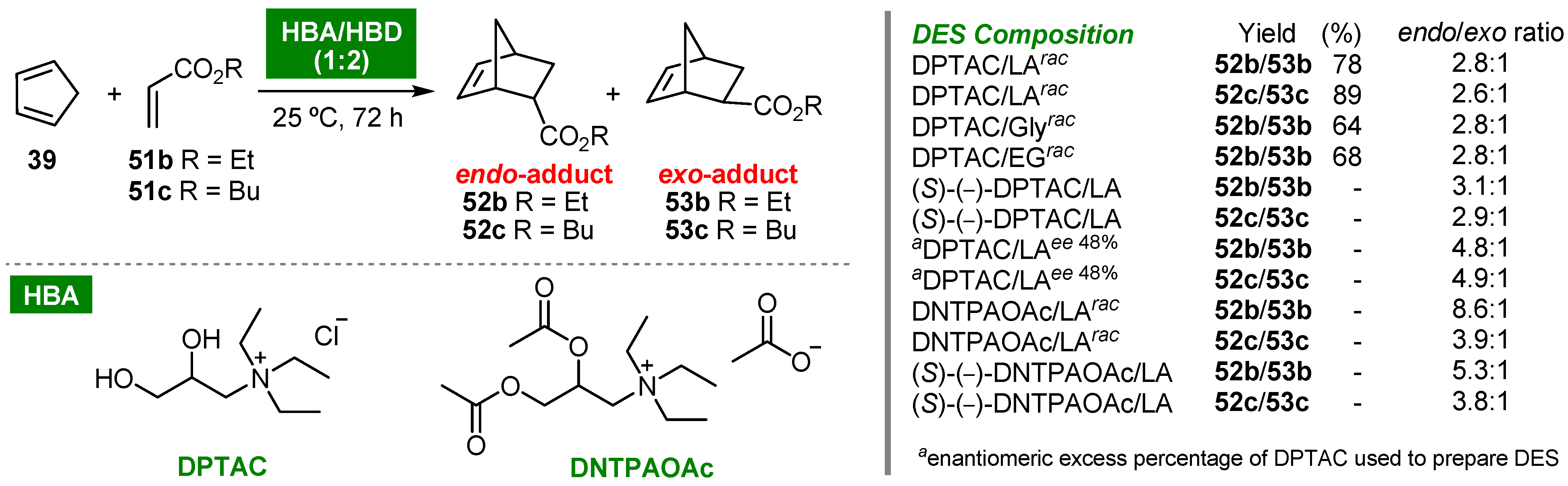
- Torres, P.; Balcells, M.; Canela-Garayoa, R. Effect of novel deep eutectic solvents on the endo/exo ratio of Diels–Alder reactions at room temperature. ACS Omega 2021, 6, 19392–19399. [Google Scholar] [CrossRef]
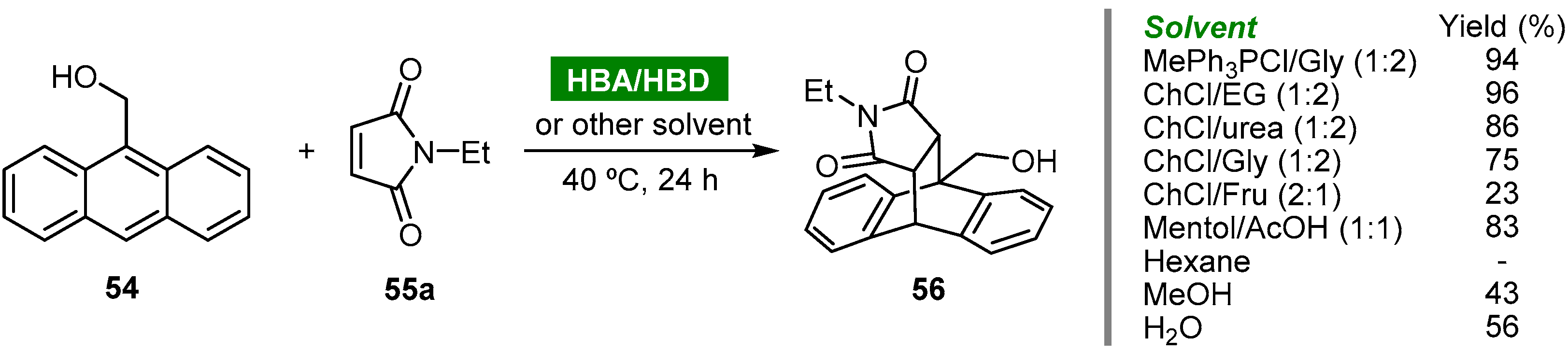
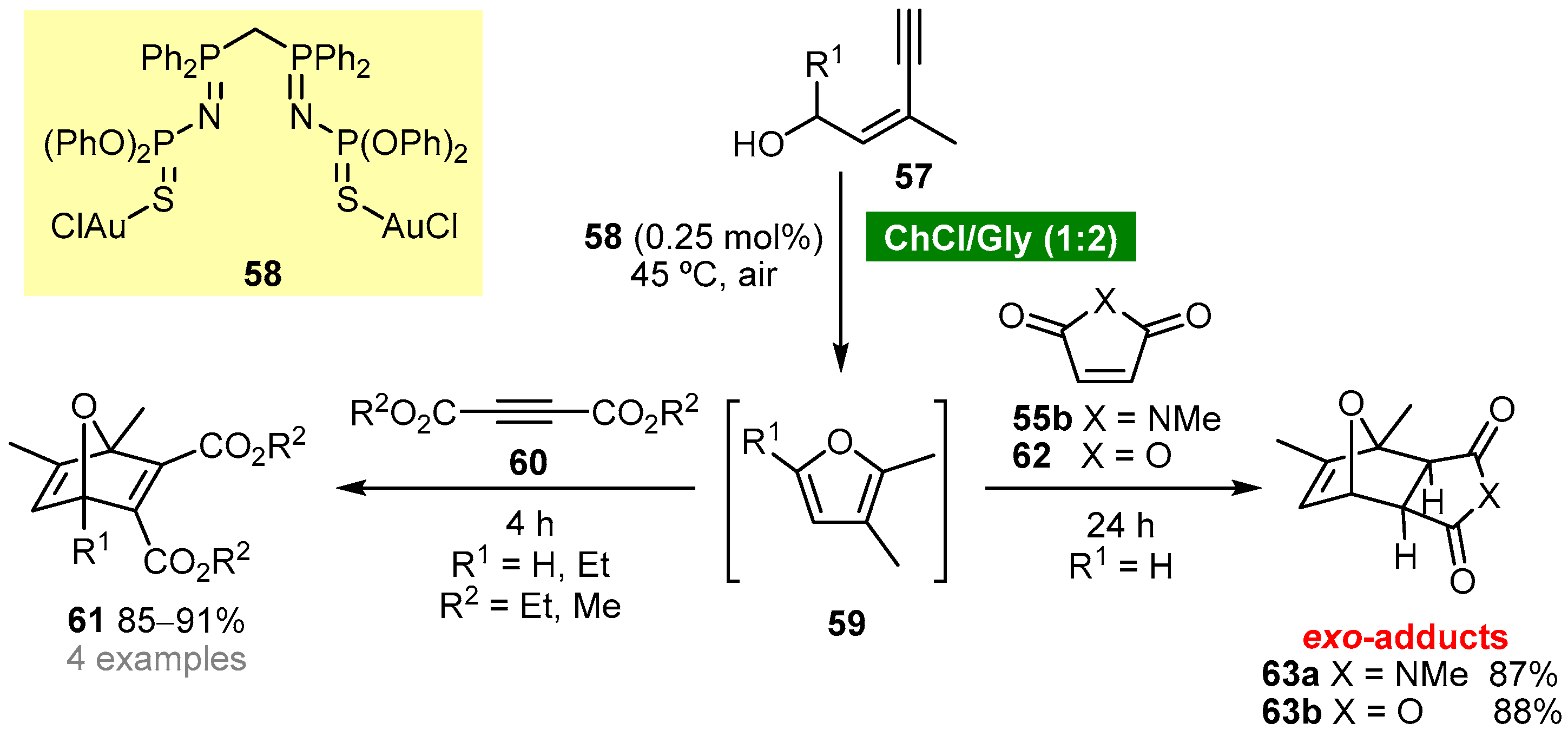
- Vidal, C.; Merz, L.; García-Álvarez, J. Deep eutectic solvents: Biorenewable reaction media for Au(I)-catalysed cycloisomerisations and one-pot tandem cycloisomerisation/Diels–Alder reactions. Green Chem. 2015, 17, 3870–3878. [Google Scholar] [CrossRef]
- Hu, Y.C.; Yuan, L.; Zhang, X.L.; Zhou, H.; Wang, P.; Li, G.Y.; Wang, A.Q.; Cong, Y.; Zhang, T.; Liang, X.M.; et al. Production of 1,2-cyclohexanedicarboxylates from diacetone alcohol and fumarates. ACS Sustain. Chem. Eng. 2019, 7, 2980–2988. [Google Scholar] [CrossRef]
- Lopes, S.M.M.; Cardoso, A.L.; Lemos, A.; Pinho e Melo, T.M.V.D. Recent advances in the chemistry of conjugated nitrosoalkenes and azoalkenes. Chem. Rev. 2018, 118, 11324–11352. [Google Scholar] [CrossRef]
- Grosso, C.; Brigas, A.; de los Santos, J.M.; Palacios, F.; Lemos, A.; Pinho e Melo, T.M.V.D. Natural deep eutectic solvents in the hetero-Diels–Alder approach to bis(indolyl)methanes. Monatsh. Chem. 2019, 150, 1275–1288. [Google Scholar] [CrossRef]
- Grosso, C.; Cardoso, A.L.; Lemos, A.; Varela, J.; Rodrigues, M.J.; Custódio, L.; Barreira, L.; Pinho e Melo, T.M.V.D. Novel approach to bis(indolyl)methanes: De novo synthesis of 1-hydroxyiminomethyl derivatives with anti-cancer properties. Eur. J. Med. Chem. 2015, 93, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Cardoso, A.L.; Rodrigues, M.J.; Marques, C.; Barreira, L.; Lemos, A.; Pinho e Melo, T.M.D.V. Hetero-Diels–Alder approach to bis(indolyl)methanes. Bioorg. Med. Chem. 2017, 25, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Gómez, A.P.; Bejarano, O.R.; Kouznetsov, V.V.; Ochoa-Puentes, C. One-pot diastereoselective synthesis of tetrahydroquinolines from star anise oil in a choline chloride/zinc chloride eutectic mixture. ACS Sustain. Chem. Eng. 2019, 7, 18630–18639. [Google Scholar] [CrossRef]
- Penã-Solórzano, D.; Kouznetsov, V.V.; Ochoa-Puentes, C. Physicochemical properties of a urea/zinc chloride eutectic mixture and its improved effect on the fast and high yield synthesis of indeno[2,1-c]quinolines. New J. Chem. 2020, 44, 7987–7997. [Google Scholar] [CrossRef]
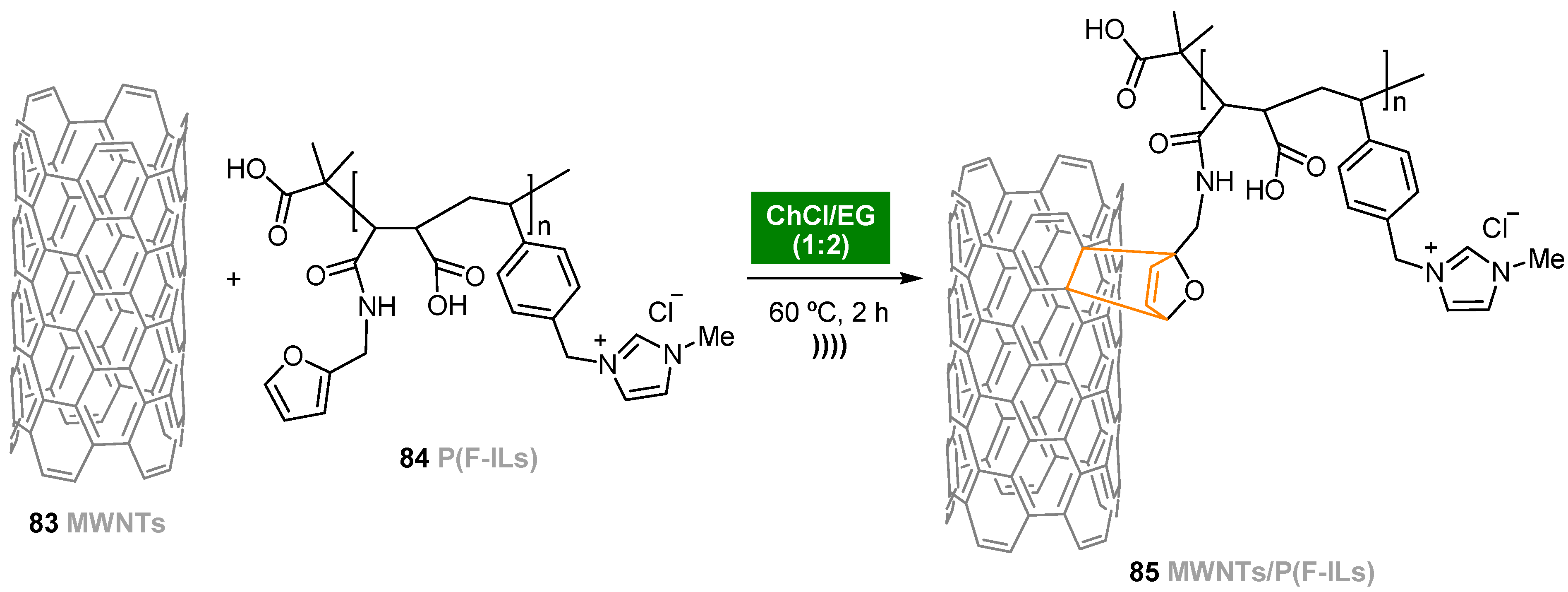
- Le, C.M.Q.; Cao, X.T.; Tu, T.T.K.; Lee, W.-K.; Lim, K.T. Facile covalent functionalization of carbon nanotubes via Diels–Alder reaction in deep eutectic solvents. Appl. Surf. Sci. 2018, 450, 122–129. [Google Scholar] [CrossRef]
- Ramesh, K.; Siboro, S.A.P.; Kim, D.W.; Lim, K.T. Ultrasound-accelerated covalent-functionalization of reduced graphene oxide with imidazolium-based poly(ionic liquid)s by Diels–Alder click reaction for supercapacitors. React. Funct. Polym. 2020, 152, 104605. [Google Scholar] [CrossRef]
- Eckert, C.A.; Knutson, B.L.; Debenedetti, P.G. Supercritical fluids as solvents for chemical and materials processing. Nature 1996, 383, 313–318. [Google Scholar] [CrossRef]
- Peach, J.; Eastoe, J. Supercritical carbon dioxide: A solvent like no other. Beilstein J. Org. Chem. 2014, 10, 1878–1895. [Google Scholar] [CrossRef] [Green Version]
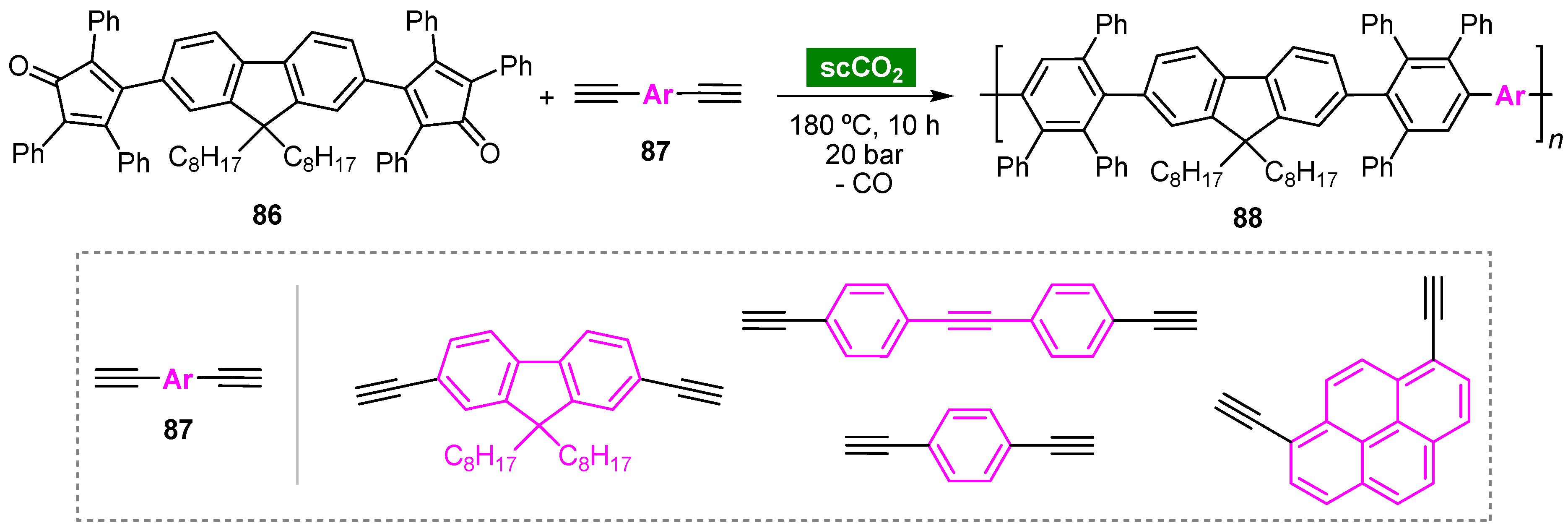
- Keshtov, M.L.; Mal’tsev, E.I.; Lopatin, A.M.; Nikitin, L.N.; Marochkin, D.V.; Perevalov, V.P.; Petrovskii, P.V.; Khokhlov, A.R. Photoluminescent phenylated polyfluorenes synthesized in an organic solvent and supercritical carbon dioxide. Polym. Sci. Ser. B 2012, 54, 106–114. [Google Scholar] [CrossRef]
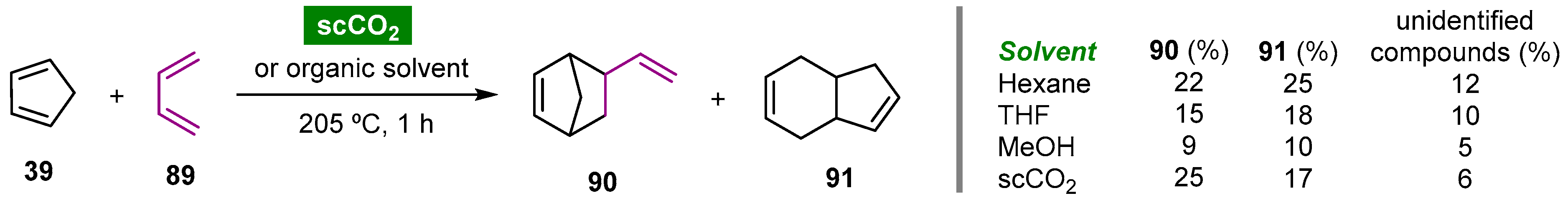
- Meng, F.-Q.; Feng, X.-J.; Wang, W.-H.; Bao, M. Synthesis of 5-vinyl-2-norbornene through Diels–Alder reaction of cyclopentadiene with 1,3-butadiene in supercritical carbon dioxide. Chin. Chem. Lett. 2017, 28, 900–904. [Google Scholar] [CrossRef]
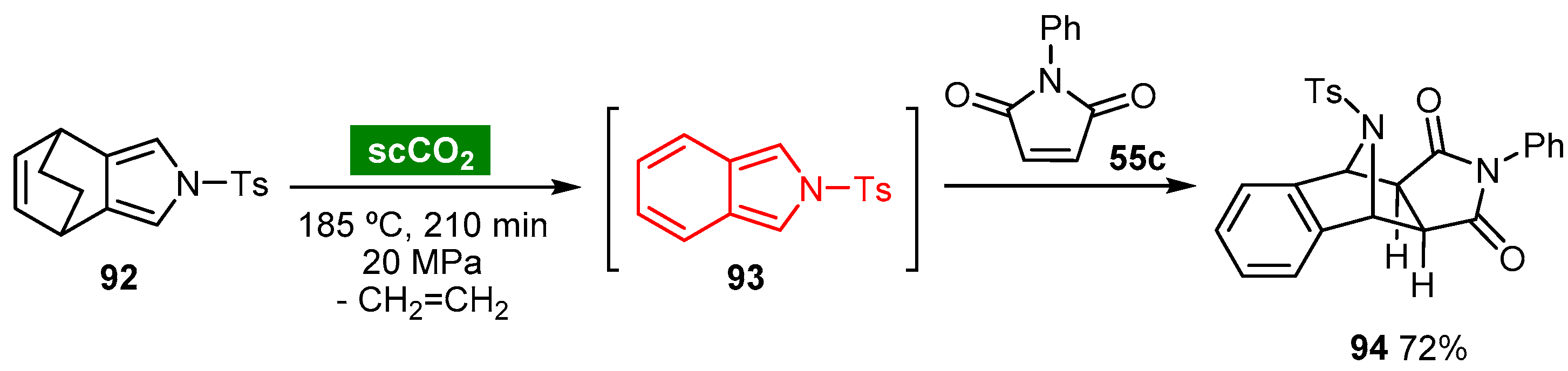
- Ito, S.; Akaki, M.; Shinozaki, Y.; Iwabe, Y.; Furuya, M.; Tobata, M.; Roppongi, M.; Sato, T.; Itoh, N.; Oba, T. Efficient synthesis of isoindoles using supercritical carbon dioxide. Tetrahedron Lett. 2017, 58, 1338–1342. [Google Scholar] [CrossRef]
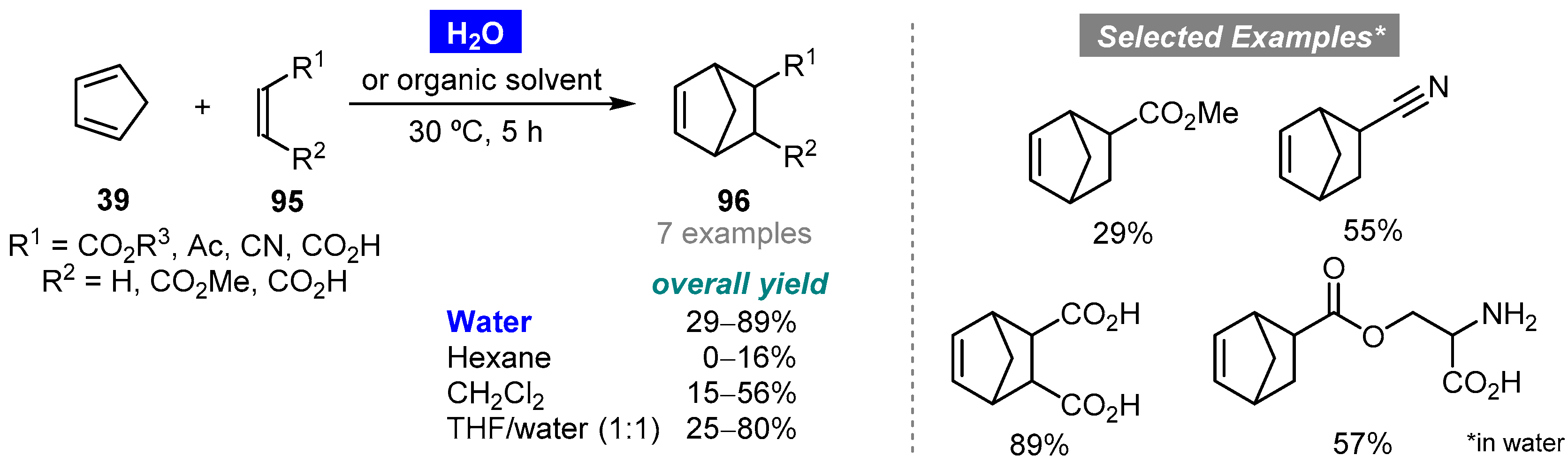
- Rideout, D.C.; Breslow, R. Hydrophobic acceleration of Diels–Alder reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar] [CrossRef]
- Breslow, R.; Maitra, U. On the origin of product selectivity in aqueous Diels–Alder reactions. Tetrahedron Lett. 1984, 25, 1239–1240. [Google Scholar] [CrossRef]
- Breslow, R.; Maitra, U.; Rideout, D. Selective Diels–Alder reactions in aqueous solutions and suspensions. Tetrahedron Lett. 1983, 24, 1901–1904. [Google Scholar] [CrossRef]
- Grieco, P.A.; Garner, P.; He, Z. Micellar catalysis in the aqueous intermolecular Diels–Alder reaction: Rate acceleration and enhanced selectivity. Tetrahedron Lett. 1983, 24, 1897–1900. [Google Scholar] [CrossRef]
- Shrinidhi, A. Diels–Alder reaction with hydrophilic dienes and dienophiles. ChemistrySelect 2016, 1, 3016–3021. [Google Scholar] [CrossRef]
- Soto-Delgado, J.; Aizman, A.; Contreras, R.; Domingo, L.R. On the catalytic effect of water in the intramolecular Diels–Alder reaction of quinone systems: A theoretical study. Molecules 2012, 17, 13687–13703. [Google Scholar] [CrossRef]
- Gallardo-Fuentes, S.; Lezana, N.; Lühr, S.; Galdámez, A.; Vilches-Herrera, M. Influence of non-covalent interactions in the exo- and regioselectivity of aza-Diels–Alder reactions: Experimental and DFT calculations. J. Org. Chem. 2019, 84, 10825–10831. [Google Scholar] [CrossRef]
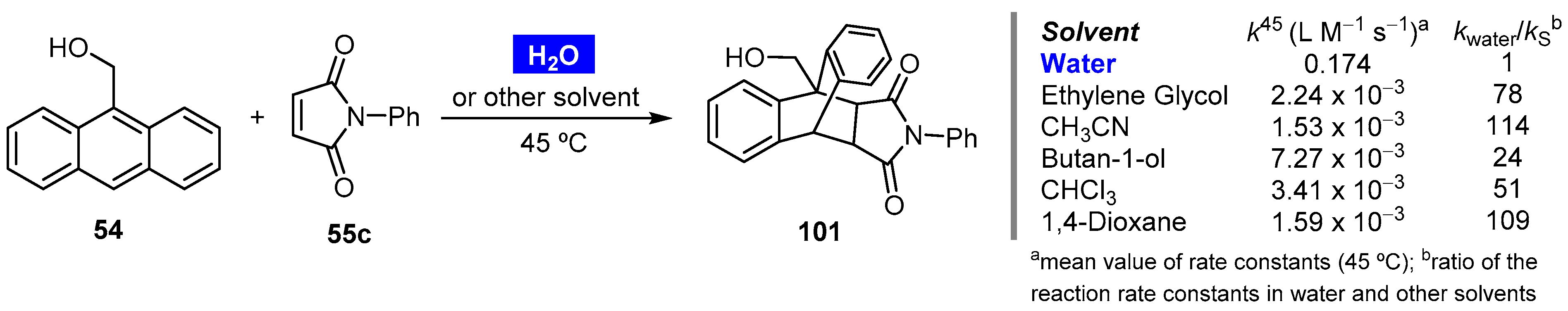
- Kiselev, V.D.; Kashaeva, H.A.; Potapova, L.N.; Kornilov, D.A.; Latypova, L.I.; Konovalov, A.I. Hydrophobic acceleration in the Diels–Alder reaction of 9-hydroxymethylanthracene with N-phenylmaleimide. Russ. Chem. Bull. 2016, 65, 2202–2205. [Google Scholar] [CrossRef]
- Nagare, A.S.; Manna, A.; Sonawane, P.D.; Kumar, A. The water-promoted Diels–Alder reaction in quaternary ammonium salts. J. Phys. Org. Chem. 2015, 28, 665–673. [Google Scholar] [CrossRef]
- Reid, J.E.S.J.; Aquino, P.H.G.; Walker, A.J.; Karadakov, P.B.; Shimizu, S. Statistical thermodynamics unveils how ions influence an aqueous Diels–Alder reaction. ChemPhysChem 2019, 20, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Tanaka, H.; Hirata, F. Theoretical study of salt effects on the Diels–Alder reaction of cyclopentadiene with methyl vinyl ketone using RISM-SCF theory. J. Phys. Chem. B 2013, 117, 14115–14121. [Google Scholar] [CrossRef] [PubMed]
- Nagare, A.S.; Manna, A.; Kumar, A. Can a Diels–Alder reaction be accelerated in a supersaturated solvent at room temperature? New J. Chem. 2016, 40, 8355–8363. [Google Scholar] [CrossRef]
- Gil, M.V.; Luque-Agudo, V.; Román, E.; Serrano, J.A. Expeditious ‘on-water’ cycloaddition between N-substituted maleimides and furans. Synlett 2014, 25, 2179–2183. [Google Scholar]
- Higson, S.; Subrizi, F.; Sheppard, T.D.; Hailes, H.C. Chemical cascades in water for the synthesis of functionalized aromatics from furfurals. Green Chem. 2016, 18, 1855–1858. [Google Scholar] [CrossRef] [Green Version]
- Kucherov, F.A.; Galkin, K.I.; Gordeev, E.G.; Ananikov, V.P. Efficient route for the construction of polycyclic systems from bioderived HMF. Green Chem. 2017, 19, 4858–4864. [Google Scholar] [CrossRef] [Green Version]
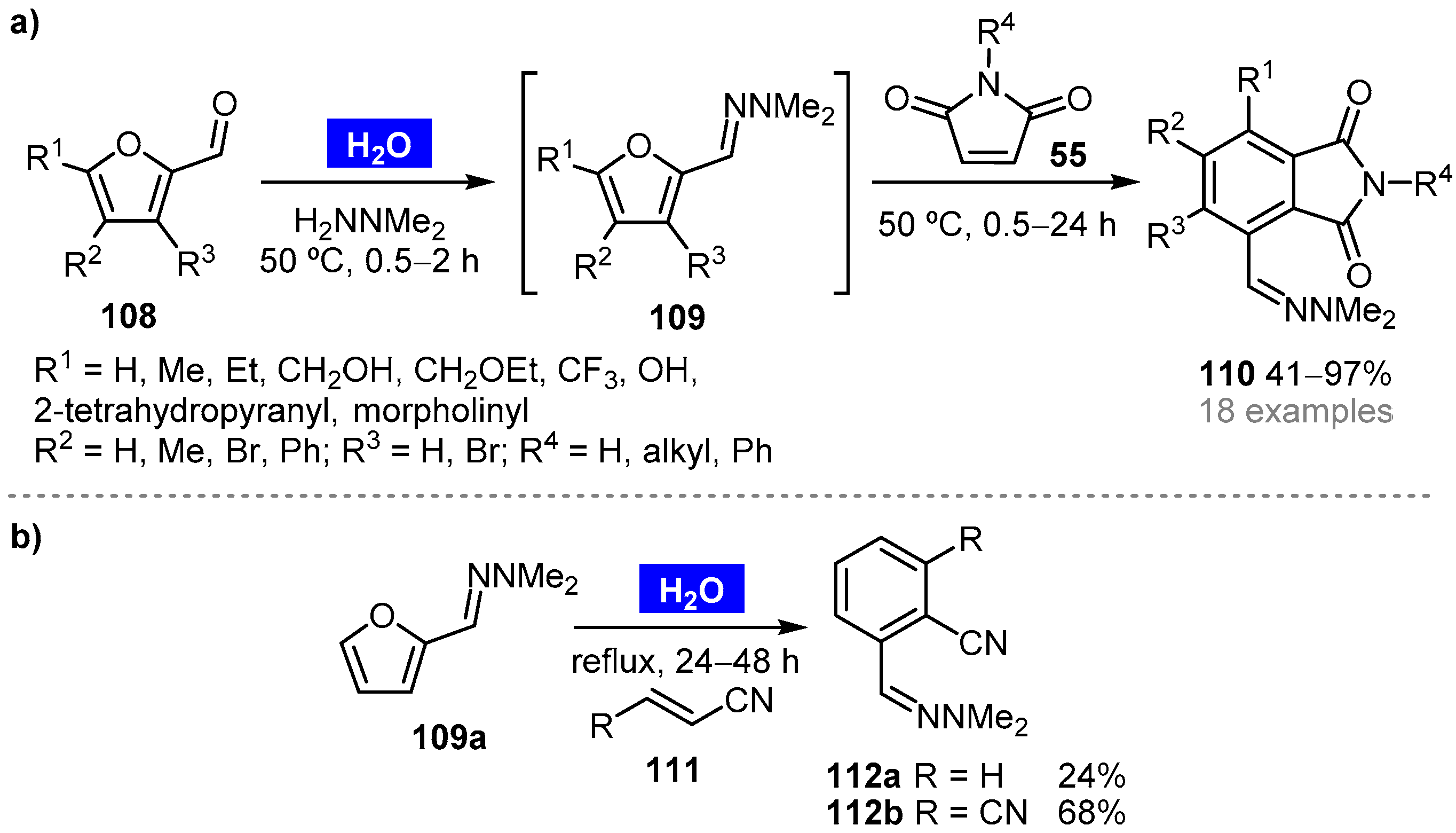
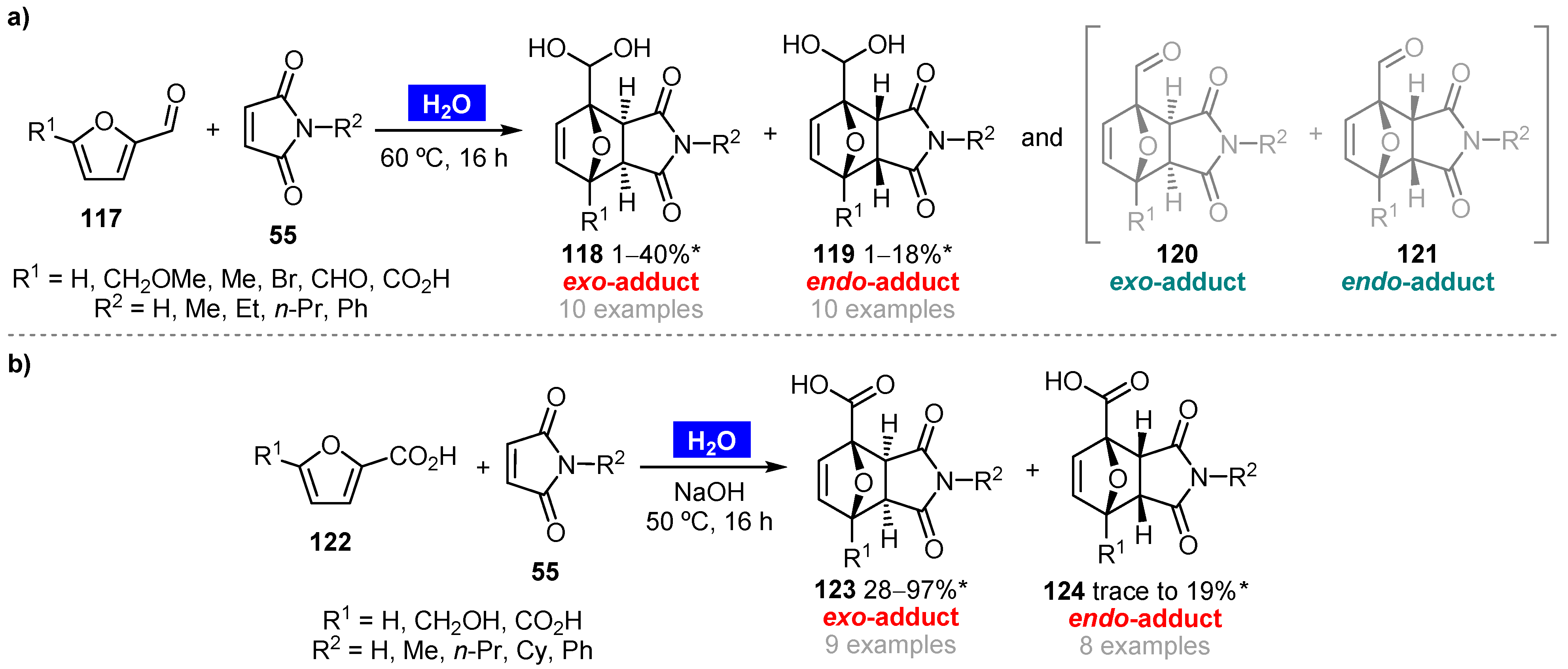
- Cioc, R.C.; Lutz, M.; Pidko, E.A.; Crockatt, M.; van der Waal, J.C.; Bruijnincx, P.C.A. Direct Diels–Alder reactions of furfural derivatives with maleimides. Green Chem. 2021, 23, 367–373. [Google Scholar] [CrossRef]
- Cioc, R.C.; Smak, T.J.; Crockatt, M.; van der Waal, J.C.; Bruijnincx, P.C.A. Furoic acid and derivatives as atypical dienes in Diels–Alder reactions. Green Chem. 2021, 23, 5503–5510. [Google Scholar] [CrossRef]
- Pinto, J.; Silva, V.L.M.; Silva, A.M.G.; Silva, A.M.S.; Costa, J.C.S.; Santos, L.M.N.B.F.; Enes, R.; Cavaleiro, J.A.S.; Vicente, A.A.M.O.S.; Teixeira, J.A.C. Ohmic heating as a new efficient process for organic synthesis in water. Green Chem. 2013, 15, 970–975. [Google Scholar] [CrossRef]
- Cardoso, M.F.C.; Gomes, A.T.P.C.; Silva, V.L.M.; Silva, A.M.S.; Neves, M.G.P.M.S.; da Silva, F.C.; Ferreira, V.F.; Cavaleiro, J.A.S. Ohmic heating assisted synthesis of coumarinyl porphyrin derivatives. RSC Adv. 2015, 5, 66192–66199. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Cui, L.; Wang, J.; Fan, X. Catalyst-free synthesis of diversely substituted 6H-benzo[c]chromenes and 6H-benzo[c]chromen-6-ones in aqueous media under MWI. Green Chem. 2012, 14, 3429–3435. [Google Scholar] [CrossRef]
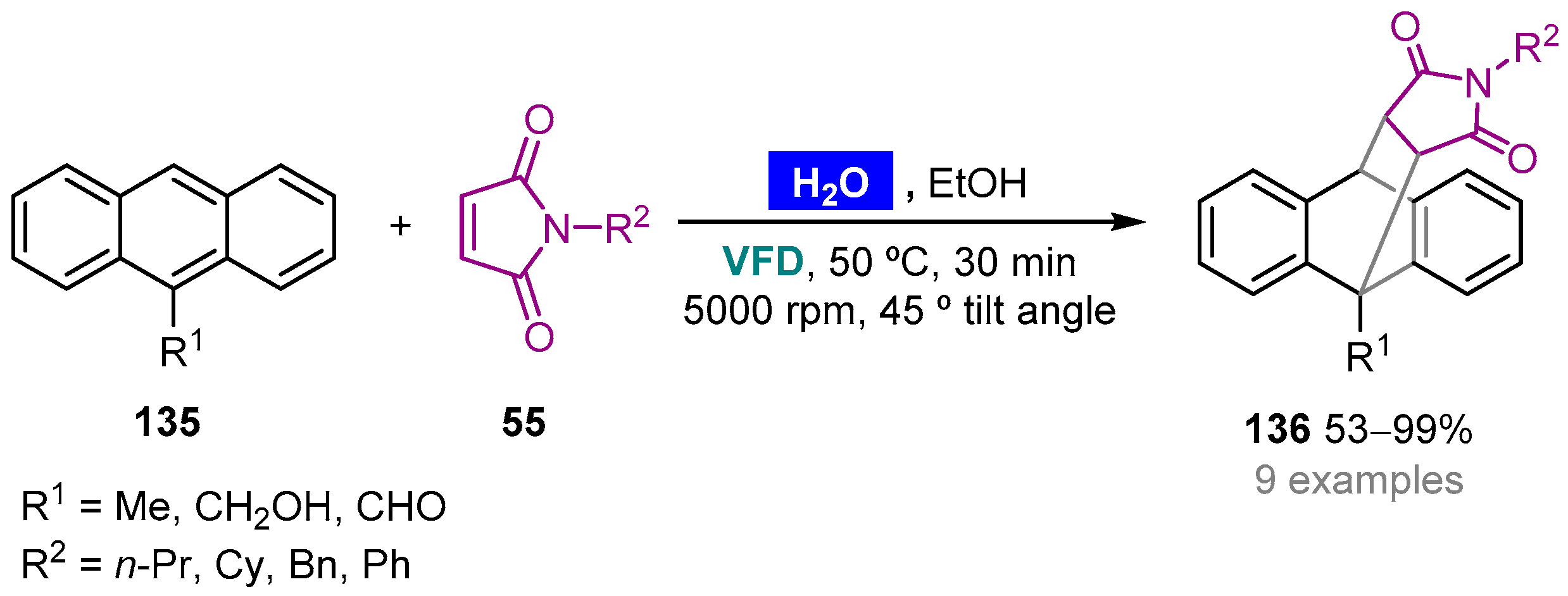
- Yasmin, L.; Stubbs, K.A.; Raston, C.L. Vortex fluidic promoted Diels–Alder reactions in an aqueous medium. Tetrahedron Lett. 2014, 55, 2246–2248. [Google Scholar] [CrossRef]
- Pereira, N.A.M.; Lopes, S.M.M.; Lemos, A.; Pinho e Melo, T.M.V.D. On-water synthesis of dipyrromethanes via bis-hetero-Diels–Alder reaction of azo- and nitrosoalkenes with pyrrole. Synlett 2014, 25, 423–427. [Google Scholar] [CrossRef]
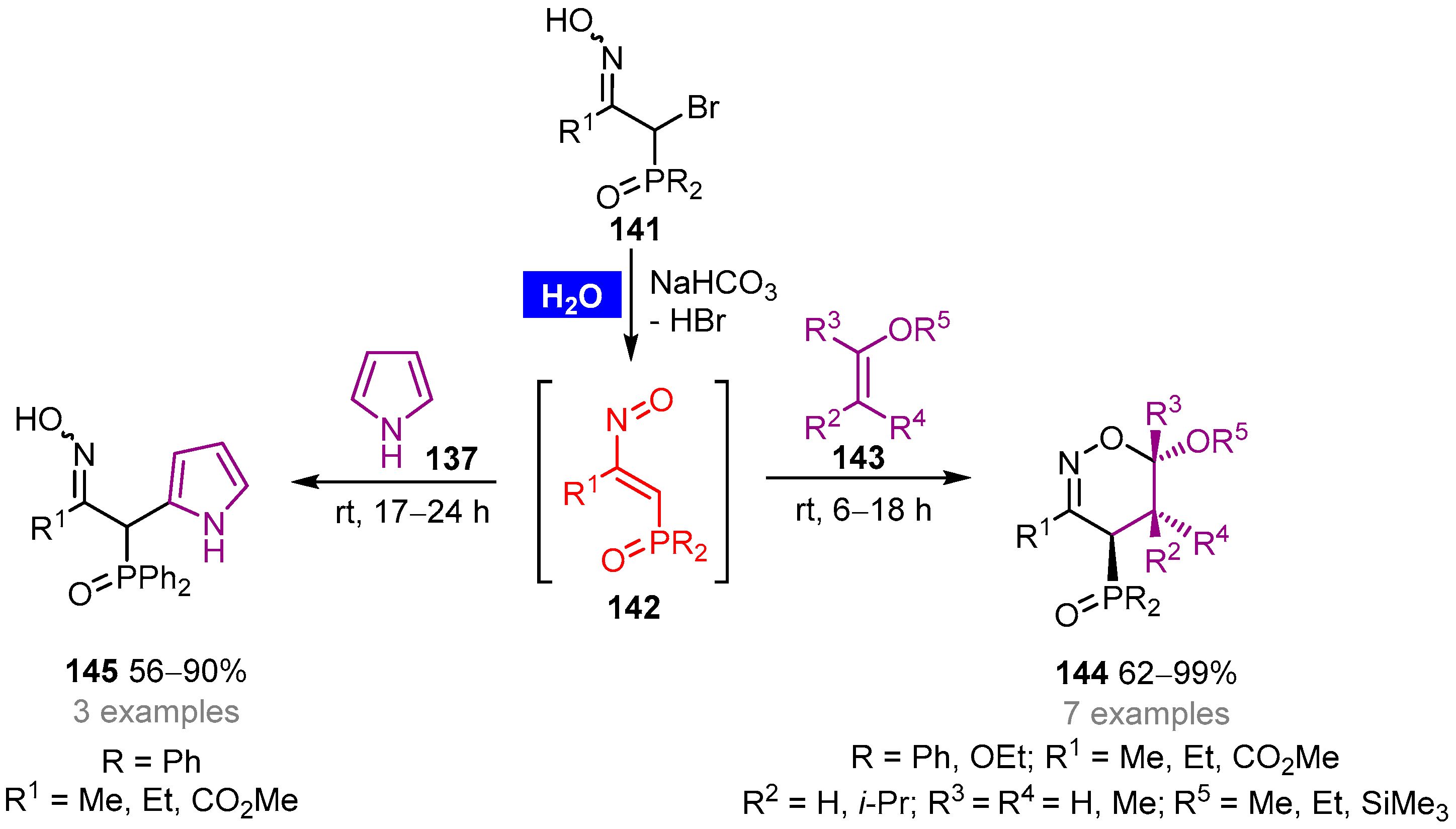
- de los Santos, J.M.; Ignacio, R.; Sbai, Z.E.; Aparicio, D.; Palacios, F. Hetero-Diels–Alder reaction of phosphorylated nitroso alkenes with enol ethers on water: A clean approach toward 1,2-oxazine derivatives. J. Org. Chem. 2014, 79, 7607–7615. [Google Scholar] [CrossRef] [PubMed]
- de los Santos, J.M.; Rubiales, G.; Sbai, Z.E.; de Retana, A.M.O.; Palacios, F. Reaction of phosphinylated nitrosoalkenes with electron-rich heterocycles. Electrophilic aromatic substitution vs. cycloaddition. Org. Biom. Chem. 2017, 15, 662–671. [Google Scholar] [CrossRef] [PubMed]
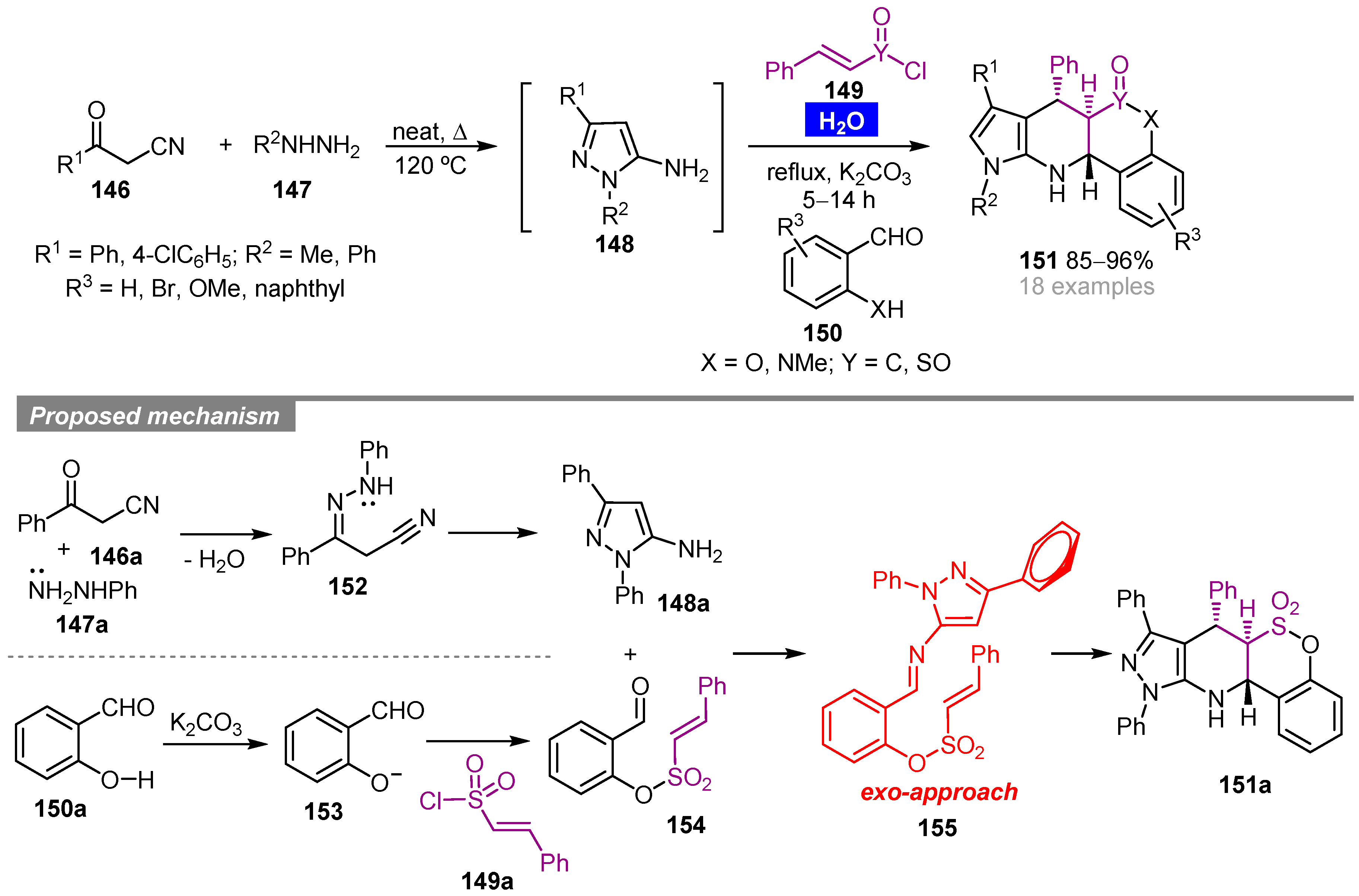
- Nazeri, M.T.; Javanbakht, S.; Shaabani, A.; Khavasi, H.R. Chemo- and diastereoselective synthesis of pyrazolo-tetrahydropyridines via multicomponent sequential aza-Diels–Alder reactions in water. ChemistrySelect 2019, 4, 14271–14275. [Google Scholar] [CrossRef]
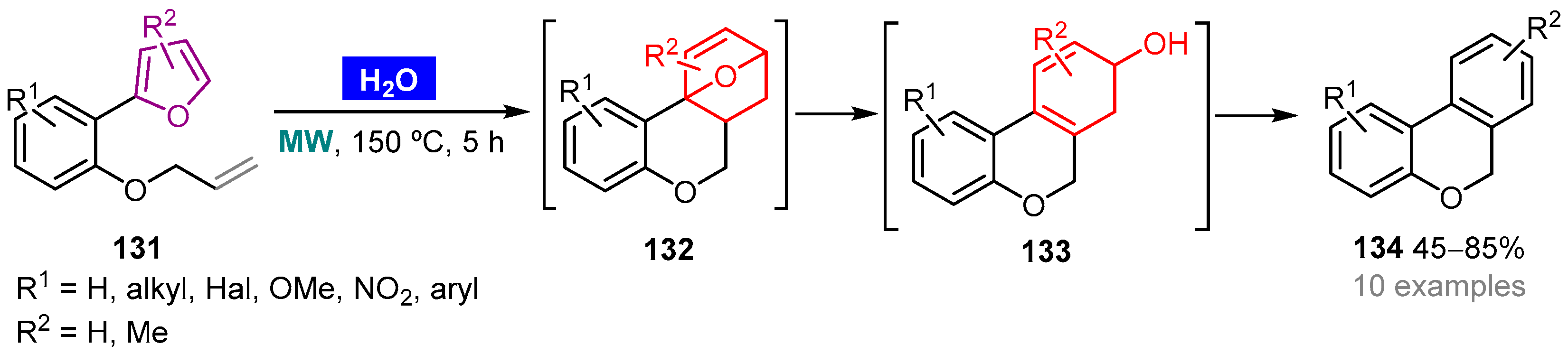
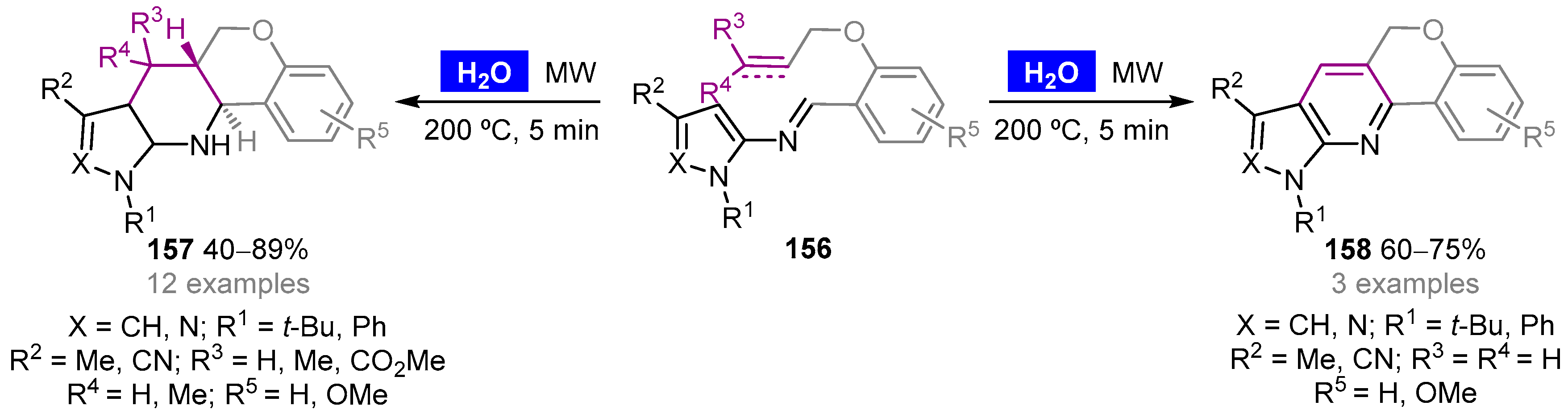
- Lezana, N.; Matus-Pérez, M.; Gáldamez, A.; Lühr, S.; Vilches-Herrera, M. Highly stereoselective and catalyst-free synthesis of annulated tetrahydropyridines by intramolecular imino-Diels–Alder reaction under microwave irradiation in water. Green Chem. 2016, 18, 3712–3717. [Google Scholar] [CrossRef]
- Ma, N.; Jiang, B.; Zhang, G.; Tu, S.-J.; Wever, W.; Li, G. New multicomponent domino reactions (MDRs) in water: Highly chemo-, regio- and stereoselective synthesis of spiro{[1,3]dioxanopyridine}-4,6-diones and pyrazolo[3,4-b] pyridines. Green Chem. 2010, 12, 1357–1361. [Google Scholar] [CrossRef]
- Jiang, B.; Ma, N.; Wang, X.-H.; Tu, S.-J.; Li, G. Microwave-assisted multicomponent reaction in water: Highly stereoselective synthesis of pyrimidinespiroisoxazolo[5,4-b]pyridine derivatives. Heterocycles 2012, 84, 765–774. [Google Scholar] [CrossRef]
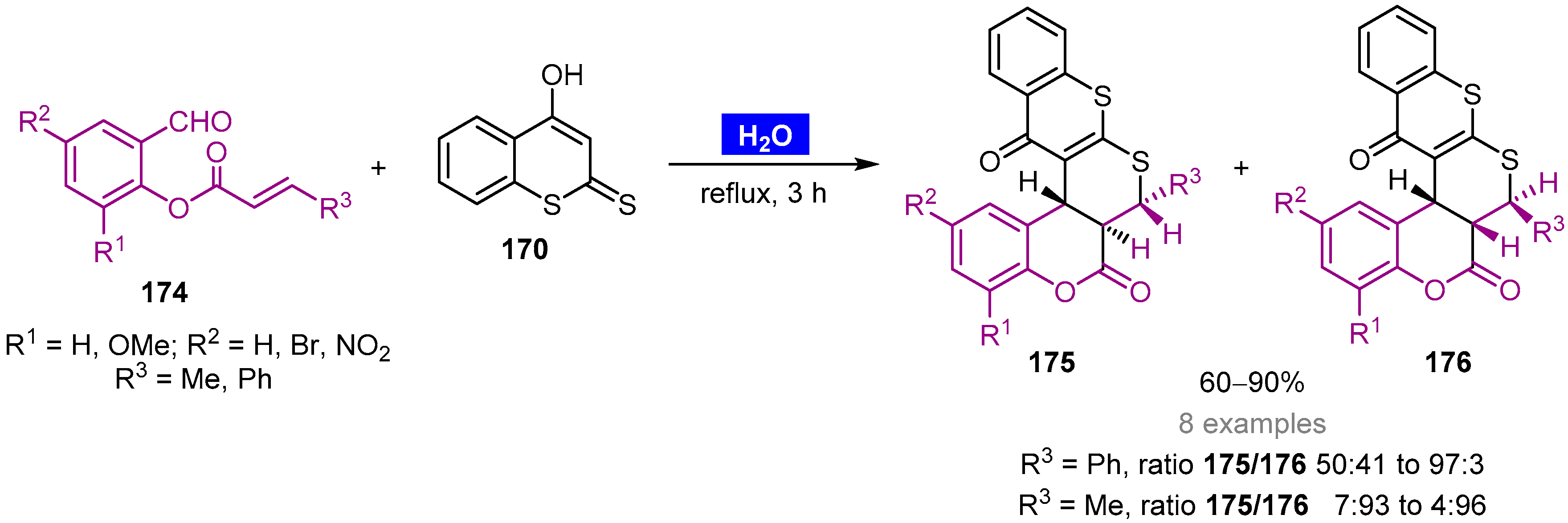
- Majumdar, K.C.; Taher, A.; Ponra, S. Catalyst-free domino-Knoevenagel-hetero-Diels–Alder reaction of terminal alkynes in water: An efficient one-step synthesis of indole-annulated thiopyranobenzopyran derivatives. Tetrahedron Lett. 2010, 51, 147–150. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Taher, A.; Ponra, S. Catalyst-free regioselective synthesis of benzopyran-annulated thiopyrano[2,3-b]thiochromen-5-(4H)-one derivatives by domino-Knoevenagel-hetero-Diels–Alder reaction of terminal alkynes with 4-hydroxy dithiocoumarin in aqueous medium. Tetrahedron Lett. 2010, 51, 2297–2300. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Taher, A.; Ponra, S. Green synthesis of benzopyran-annulated thiopyrano[2,3-b] thiochromen-5(4H)-ones by domino Knoevenagel-hetero-Diels–Alder reaction. Synthesis 2010, 23, 4043–4050. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Kiamehr, M.; Taheri, S.; Mirjafary, Z. Synthesis of novel polycyclic indole-annulated thiopyranocoumarin derivatives via domino Knoevenagel-hetero-Diels–Alder reaction in aqueous media. Helv. Chim. Acta 2010, 93, 964–973. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Kiamehr, M.; Khodabakhshi, M.R.; Mirjafary, Z.; Fathi, S.; Saeidian, H. A new domino Knoevenagel-hetero-Diels–Alder reaction: An efficient catalyst-free synthesis of novel thiochromone-annulated thiopyranocoumarin derivatives in aqueous medium. Tetrahedron 2010, 66, 8615–8622. [Google Scholar] [CrossRef]
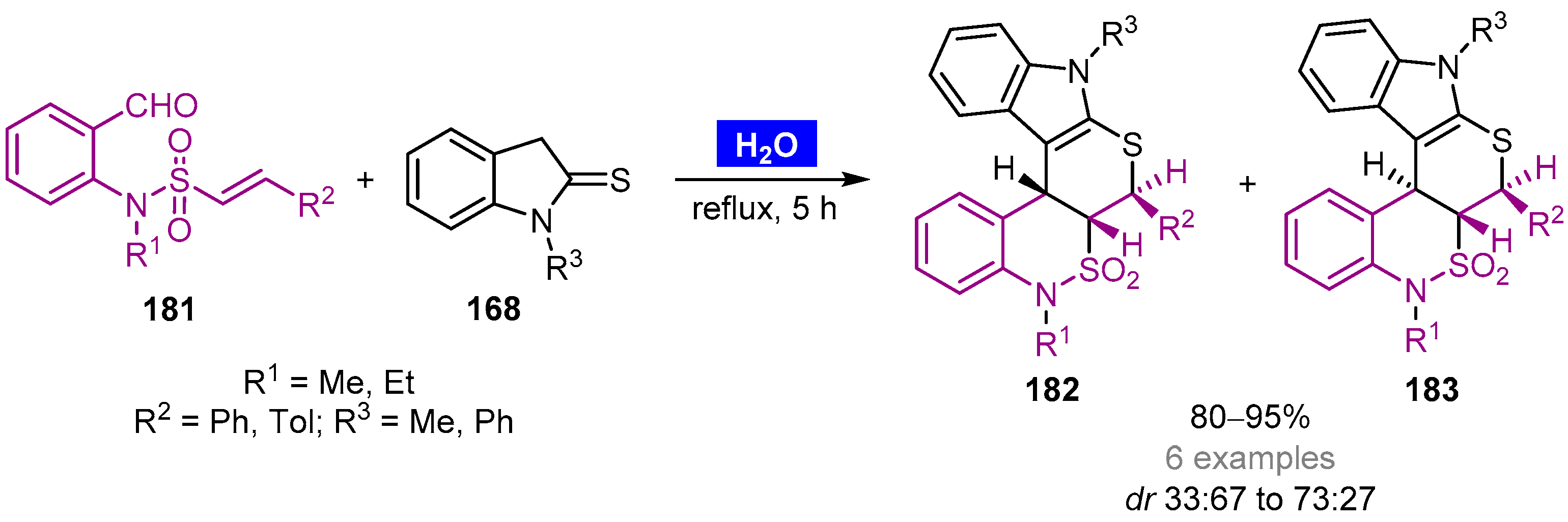
- Ghandi, M.; Mohammadimehr, E.; Sadeghzadeh, M.; Bozcheloei, A.H. Efficient access to novel hexahydro-chromene and tetrahydro-pyrano[2,3-d]pyrimidine-annulated benzo-δ-sultones via a domino Knoevenagel-hetero-Diels–Alder reaction in water. Tetrahedron 2011, 67, 8484–8491. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Khodabakhshi, M.R.; Kiamehr, M.; Ghahremannejad, Z. Synthesis of novel pentacyclic thiopyrano indole-annulated benzo-δ-sultone derivatives via a domino Knoevenagel-hetero-Diels–Alder reaction in water. Tetrahedron Lett. 2013, 54, 2685–2689. [Google Scholar] [CrossRef]
- Kiamehr, M.; Khademi, F.; Jafari, B.; Langer, P. Efficient synthesis of pentacyclic benzosultam-annulated thiopyranoindoles via domino Knoevenagel/intramolecular hetero-Diels–Alder reactions in water. Chem. Heterocycl. Compd. 2020, 56, 392–398. [Google Scholar] [CrossRef]
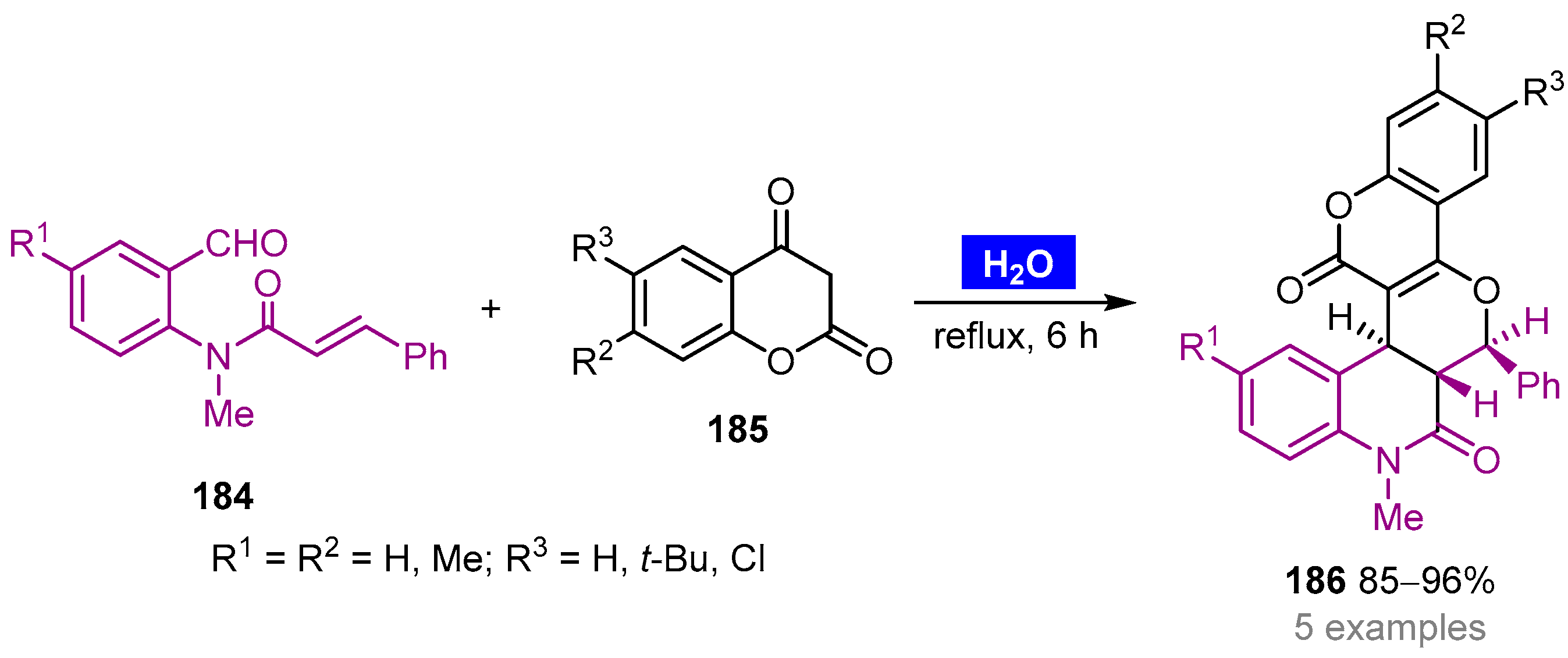
- Ghandi, M.; Feizi, S.; Nazeri, M.T.; Notash, B. Efficient access to novel tetra- and pentacyclic dihydroquinolin-2-ones by catalyst-free domino Knoevenagel hetero-Diels–Alder reactions from N-(2-formylphenyl)-N-methylcinnamamides and cyclic 1,3-dicarbonyls in water. J. Iran. Chem. Soc. 2017, 14, 177–187. [Google Scholar] [CrossRef]
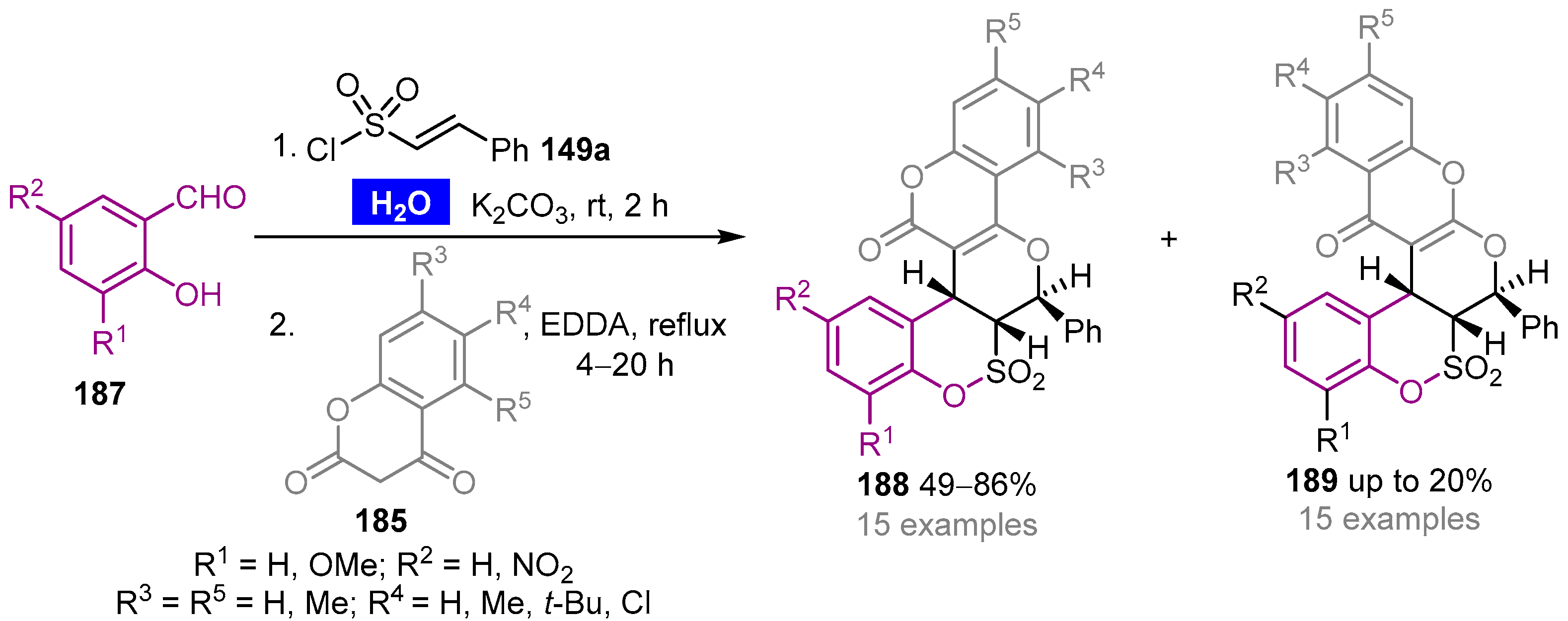
- Ghandi, M.; Nazeri, M.T.; Kubicki, M. An efficient one-pot, regio- and stereoselective synthesis of novel pentacyclic-fused pyrano[3,2,c] chromenone or quinolinone benzosultone derivatives in water. Tetrahedron 2013, 69, 4979–4989. [Google Scholar] [CrossRef]
- Palasz, A. A green approach to the synthesis of fused uracils: Pyrano[2,3-d]pyrimidines. ‘On-water’ one-pot synthesis by domino Knoevenagel/Diels–Alder reactions. Synthesis 2010, 23, 4021–4032. [Google Scholar] [CrossRef]
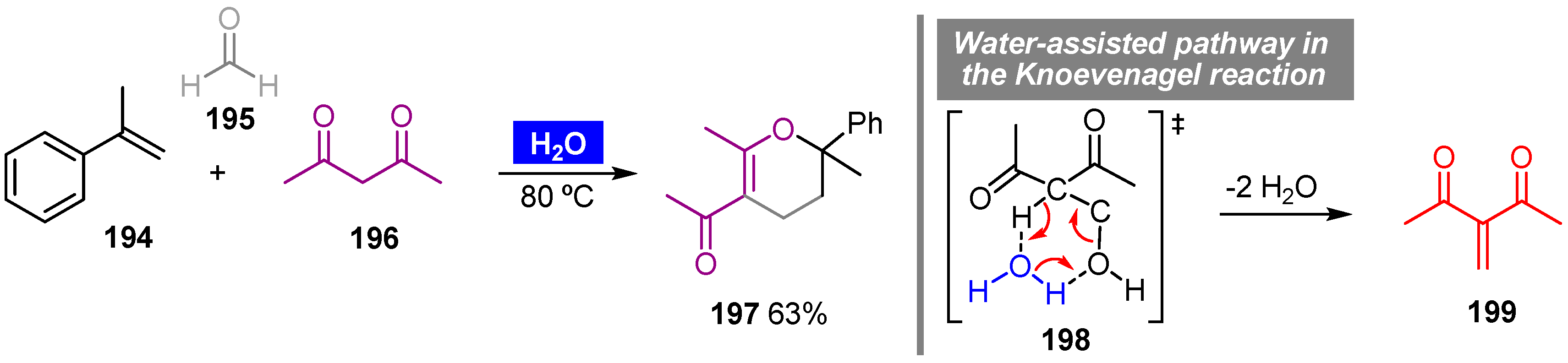
- Frapper, G.; Bachmann, C.; Gu, Y.L.; De Sousa, R.C.; Jérôme, F. Mechanisms of the Knoevenagel hetero Diels–Alder sequence in multicomponent reactions to dihydropyrans: Experimental and theoretical investigations into the role of water. Phys. Chem. Chem. Phys. 2011, 13, 628–636. [Google Scholar] [CrossRef] [PubMed]
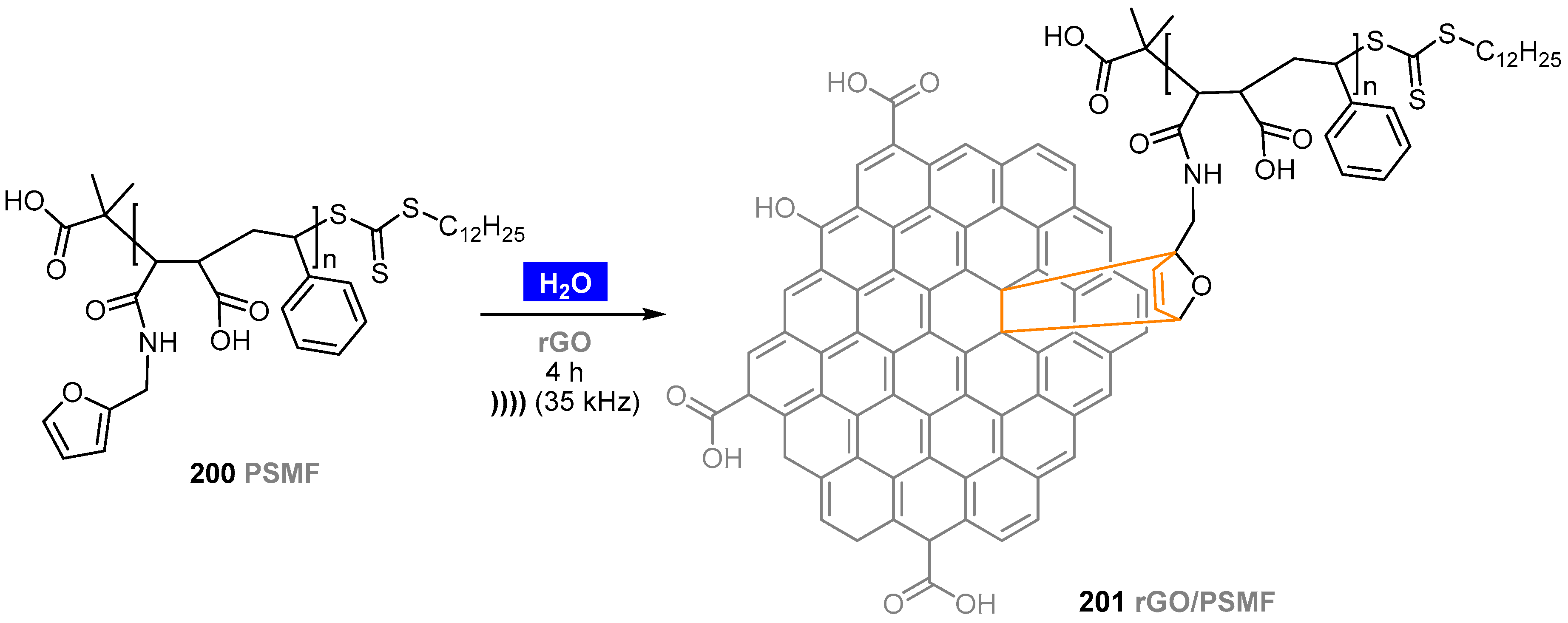
- Ramesh, K.; Anugrah, D.S.B.; Mishra, A.K.; Ahn, B.-H.; Gal, Y.-S.; Lim, K.T. Green and sono synthetic approach for direct-functionalization of reduced graphene oxide with poly(styrene-alt-maleic anhydride) by Diels Alder “click” reaction. Appl. Surf. Sci. 2020, 504, 144482. [Google Scholar] [CrossRef]
- Wei, H.L.; Yang, J.; Chu, H.J.; Yang, Z.; Ma, C.C.; Yao, K. Diels–Alder reaction in water for the straightforward preparation of thermoresponsive hydrogels. J. Appl. Polym. Sci. 2011, 120, 974–980. [Google Scholar] [CrossRef]
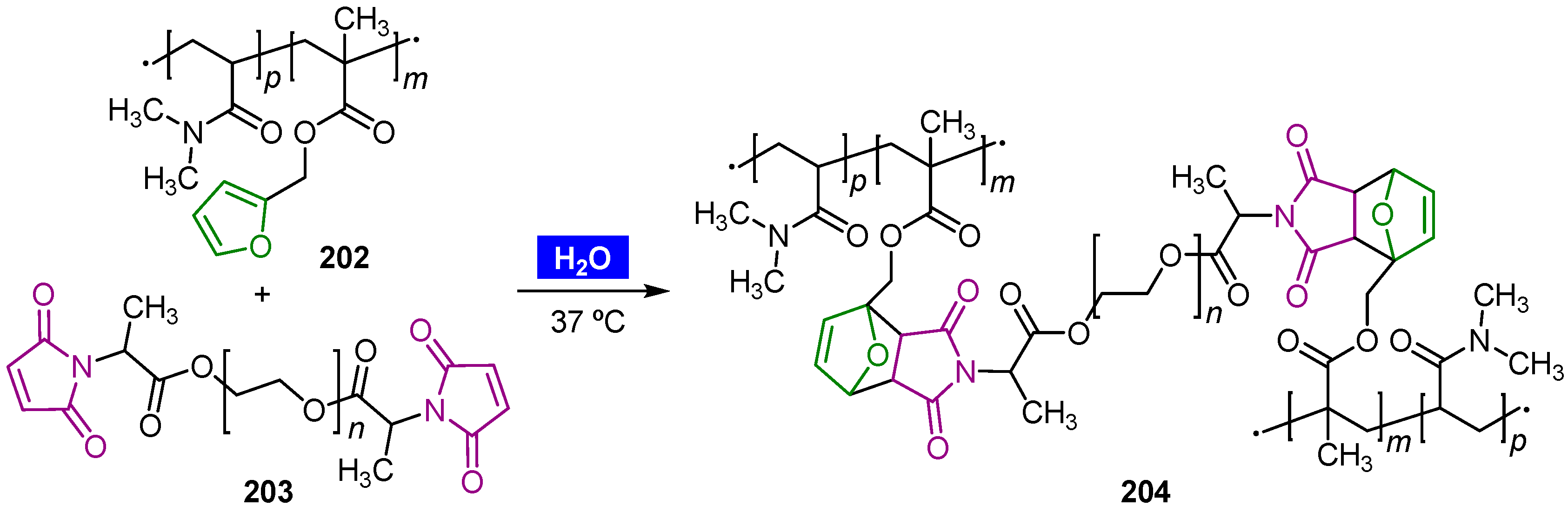
- Kirchhof, S.; Brandl, F.P.; Hammer, N.; Goepferich, A.M. Investigation of the Diels–Alder reaction as a cross-linking mechanism for degradable poly(ethylene glycol) based hydrogels. J. Mater. Chem. B 2013, 1, 4855–4864. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, C.M.; Owen, S.C.; Shoichet, M.S. Diels–Alder click cross-linked hyaluronic acid hydrogels for tissue engineering. Biomacromolecules 2011, 12, 824–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.; Rubin, J.P.; Marra, K.G. Direct synthesis of biodegradable polysaccharide derivative hydrogels through aqueous Diels–Alder chemistry. Macromol. Rapid Commun. 2011, 32, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Ma, Y.; Zhang, Z.; Mao, J.; Tan, H.; Hu, X. Biodegradable hyaluronic acid hydrogels to control release of dexamethasone through aqueous Diels–Alder chemistry for adipose tissue engineering. Mater. Sci. Eng. C 2015, 56, 311–317. [Google Scholar] [CrossRef]
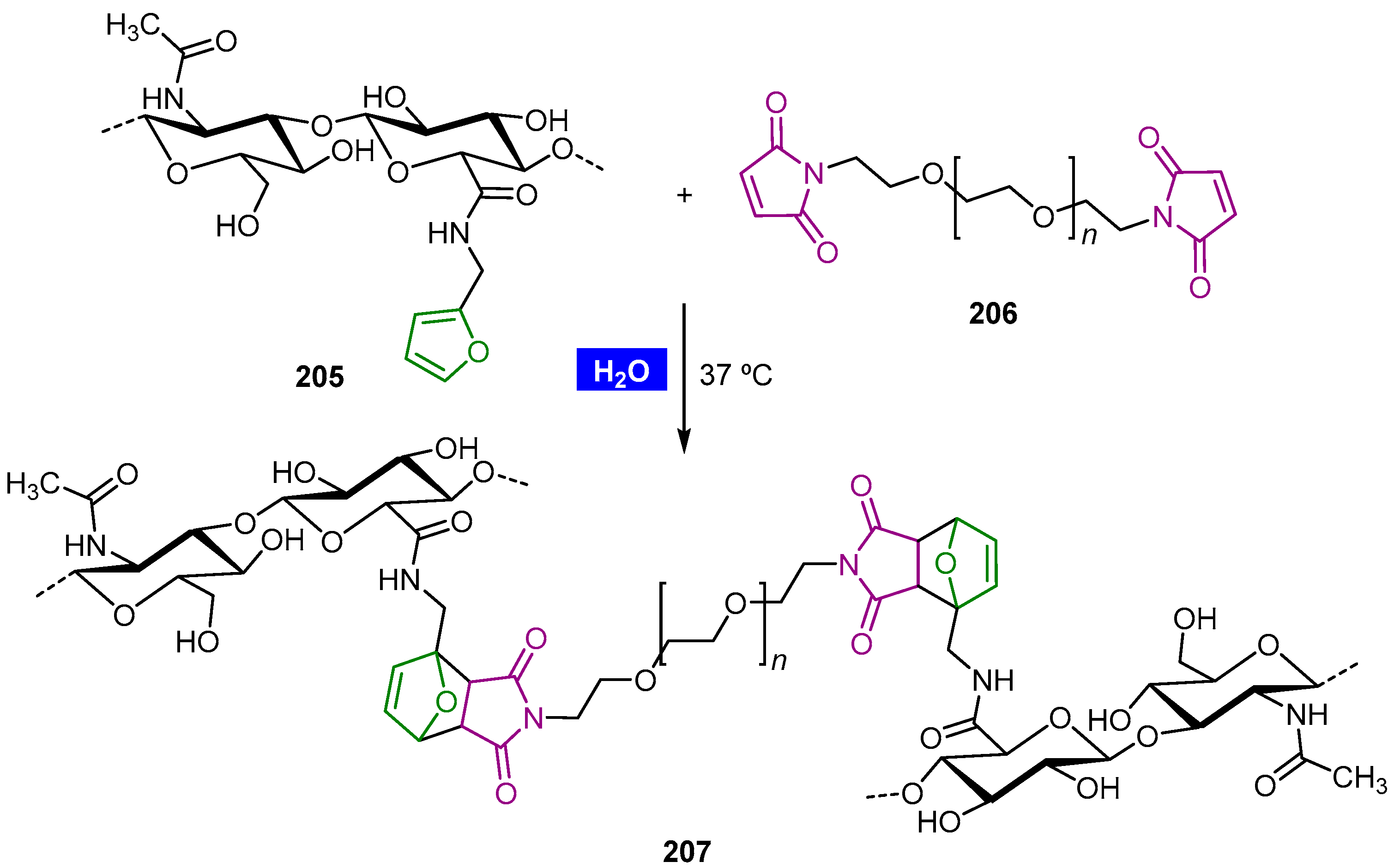
- Kramer, R.K.; Belgacem, M.N.; Carvalho, A.J.F.; Gandini, A. Thermally reversible nanocellulose hydrogels synthesized via the furan/maleimide Diels–Alder click reaction in water. Int. J. Biol. Macromol. 2019, 141, 493–498. [Google Scholar] [CrossRef]
- Li, D.-Q.; Wang, S.-Y.; Meng, Y.-J.; Guo, Z.-W.; Cheng, M.-M.; Li, J. Fabrication of self-healing pectin/chitosan hybrid hydrogel via Diels–Alder reactions for drug delivery with high swelling property, pH-responsiveness, and cytocompatibility. Carbohydr. Polym. 2021, 268, 118244. [Google Scholar] [CrossRef]
- García-Astrain, C.; Gandini, A.; Coelho, D.; Mondragon, I.; Retegi, A.; Eceiza, A.; Corcuera, M.A.; Gabilondo, N. Green chemistry for the synthesis of methacrylate-based hydrogels crosslinked through Diels–Alder reaction. Eur. Polym. J. 2013, 49, 3998–4007. [Google Scholar] [CrossRef]
- García-Astrain, C.; Algar, I.; Gandini, A.; Eceiza, A.; Corcuera, M.A.; Gabilondo, N. Hydrogel synthesis by aqueous Diels–Alder reaction between furan modified methacrylate and polyetheramine-based bismaleimides. J. Polym. Sci. A Polym. Chem. 2015, 53, 699–708. [Google Scholar] [CrossRef]
- González, K.; García-Astrain, C.; Santamaria-Echart, A.; Ugarte, L.; Avérous, L.; Eceiza, A.; Gabilondo, N. Starch/graphene hydrogels via click chemistry with relevant electrical and antibacterial properties. Carbohydr. Polym. 2018, 202, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Guaresti, O.; García-Astrain, C.; Urbina, L.; Eceiza, A.; Gabilondo, N. Reversible swelling behaviour of Diels–Alder clicked chitosan hydrogels in response to pH changes. Express Polym. Lett. 2019, 13, 27–36. [Google Scholar] [CrossRef]
- González, K.; Guaresti, O.; Palomares, T.; Alonso-Varona, A.; Eceiza, A.; Gabilondo, N. The role of cellulose nanocrystals in biocompatible starch-based clicked nanocomposite hydrogels. Int. J. Biol. Macromol. 2020, 143, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Rezende, T.R.M.; Varejão, J.O.S.; Sousa, A.L.L.A.; Castañeda, S.M.B.; Fernandes, S.A. Tetrahydroquinolines by the multicomponent Povarov reaction in water: Calix[n]arene-catalysed cascade process and mechanistic insights. Org. Biomol. Chem. 2019, 17, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Braga, I.B.; Castañeda, S.M.B.; de Assis, J.V.; Barros, A.O.; Amarante, G.W.; Valdo, A.K.S.M.; Martins, F.T.; Rosolen, A.F.P.; Pilau, E.; Fernandes, S.A. Anise essential oil as a sustainable substrate in the multicomponent double Povarov reaction for julolidine synthesis. J. Org. Chem. 2020, 85, 15622–15630. [Google Scholar] [CrossRef]
- Kaboudin, B.; Sohrabi, M.; Kazemi, F. Synthesis of julolidines via one-pot cascade three component Povarov reaction in the presence of silica sulfuric acid. J. Heterocycl. Chem. 2021, 58, 1594–1600. [Google Scholar] [CrossRef]
- Imrich, H.-G.; Conrad, J.; Bubrin, D.; Beifuss, U. From nitrobenzenes to substituted tetrahydroquinolines in a single step by a domino reduction/imine formation/aza-Diels–Alder reaction. J. Org. Chem. 2015, 80, 2319–2332. [Google Scholar] [CrossRef]
- Imrich, H.-G.; Conrad, J.; Beifuss, U. The first domino reduction/imine formation/intramolecular aza-Diels–Alder reaction for the diastereoselective preparation of tetrahydrochromano[4,3-b]quinolines. Eur. J. Org. Chem. 2016, 2016, 5706–5715. [Google Scholar] [CrossRef]
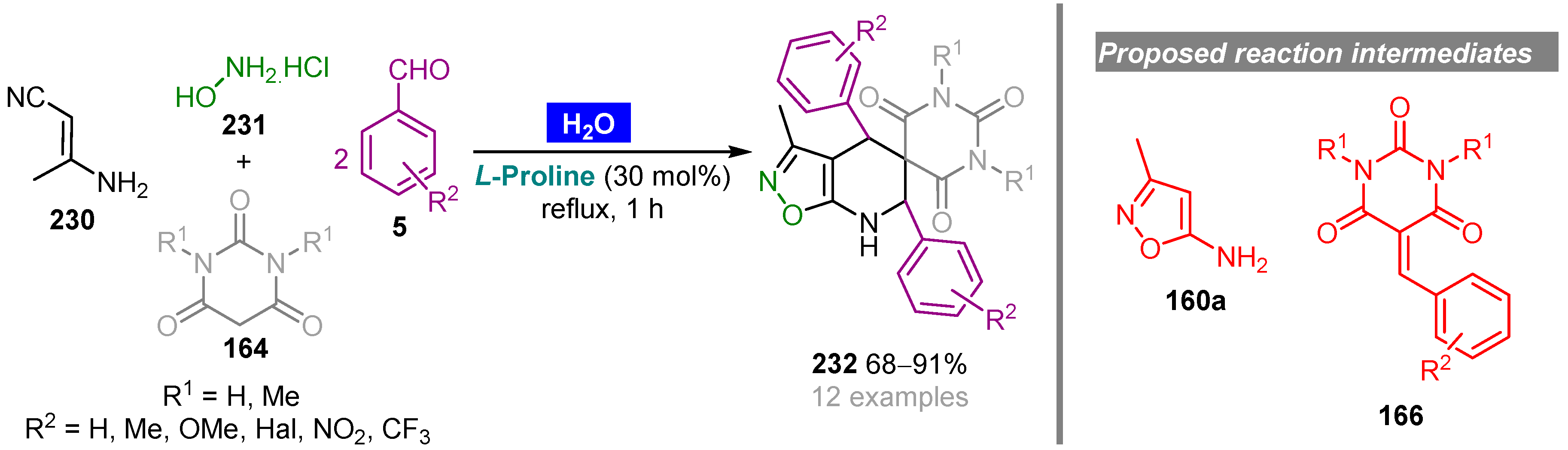
- Dommaraju, Y.; Borthakur, S.; Prajapati, D. L-Proline-catalysed one-pot aza-Diels–Alder reaction in water: Regioselective synthesis of spiro(isoxazolo[5,4-b]pyridine-5,5’-pyrimidine) derivatives. Synlett 2018, 29, 1195–1198. [Google Scholar] [CrossRef]
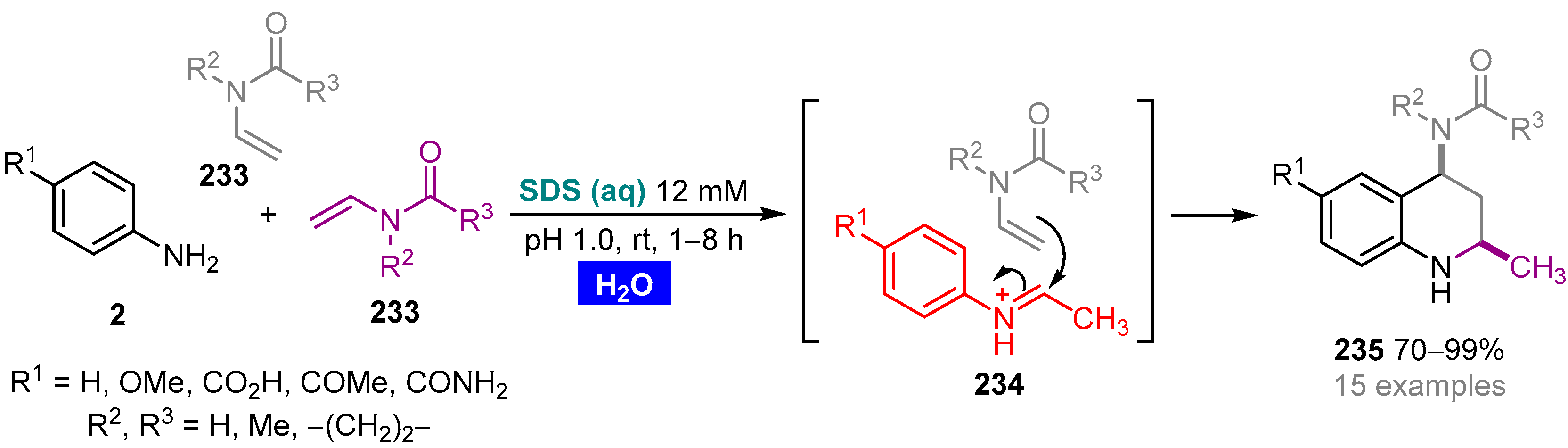
- Arenas, D.R.M.; Bonilla, C.A.M.; Kouznetsov, V.V. Aqueous SDS micelle-promoted acid-catalyzed domino ABB’ imino Diels–Alder reaction: A mild and efficient synthesis of privileged 2-methyl-tetrahydroquinoline scaffolds. Org. Biomol. Chem. 2013, 11, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
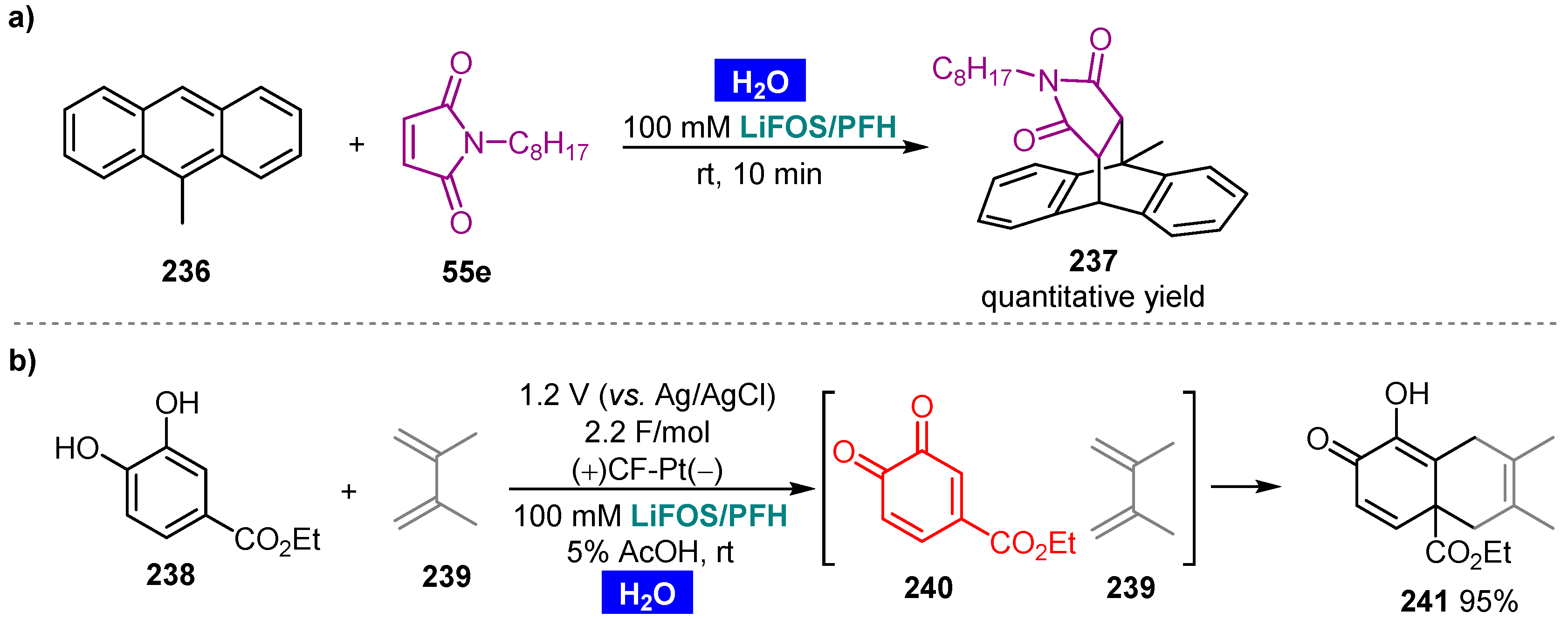
- Nishimoto, K.; Okada, Y.; Kim, S.; Chiba, K. Rate acceleration of Diels–Alder reactions utilizing a fluorous micellar system in water. Electrochim. Acta 2011, 56, 10626–10631. [Google Scholar] [CrossRef]
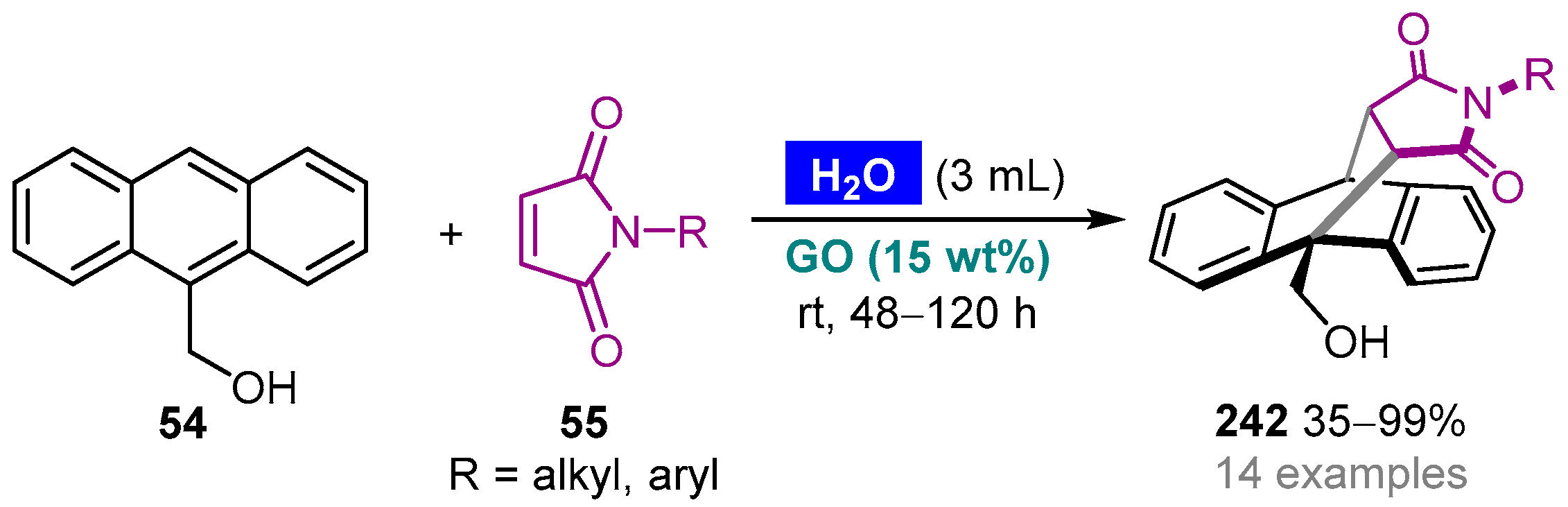
- Girish, Y.R.; Pandit, S.; Pandit, S.; De, M. Graphene oxide as a carbocatalyst for a Diels–Alder reaction in an aqueous medium. Chem. Asian J. 2017, 12, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jin, S.; Kan, Y.; Zhang, Y.J.; Zhang, W. Highly active asymmetric Diels–Alder reactions catalyzed by C-2-symmetric bipyrrolidines: Catalyst recycling in water medium and insight into the catalytic mode. Tetrahedron 2010, 66, 3849–3854. [Google Scholar] [CrossRef]
- Guizzetti, S.; Benaglia, M.; Siegel, J.S. Poly(methylhydrosiloxane)-supported chiral imidazolinones: New versatile, highly efficient and recyclable organocatalysts for stereoselective Diels–Alder cycloaddition reactions. Chem. Commun. 2012, 48, 3188–3190. [Google Scholar] [CrossRef]
- Shi, J.Y.; Wang, C.A.; Li, Z.J.; Wang, Q.; Zhang, Y.; Wang, W. Heterogeneous organocatalysis at work: Functionalization of hollow periodic mesoporous organosilica spheres with MacMillan catalyst. Chem. Eur. J. 2011, 17, 6206–6213. [Google Scholar] [CrossRef]
- Vamisetti, G.B.; Chowdhury, R.; Kumar, M.; Ghoshi, S.K. “On water” organocatalyzed [4+2] cycloaddition of enones and nitro dienes for the enantioselective synthesis of densely substituted cyclohexanones. Org. Lett. 2016, 18, 1964–1967. [Google Scholar] [CrossRef]
- Vamisetti, G.B.; Chowdhury, R.; Kumar, M.; Ghosh, S.K. ‘On water’ organocatalyzed enantioselective synthesis of highly functionalized cyclohexanones with an all-carbon quaternary centre from allylidene malononitriles and enones. Tetrahedron Asymmetry 2017, 28, 317–323. [Google Scholar] [CrossRef]
- Park, S.; Ikehata, K.; Sugiyama, H. Solid-supported DNA for asymmetric synthesis: A stepping-stone toward practical applications. Biomater. Sci. 2013, 1, 1034–1036. [Google Scholar] [CrossRef]
- Albada, H.B.; Rosati, F.; Coquière, D.; Roelfes, G.; Liskamp, R.M.J. Enantioselective CuII-catalyzed Diels–Alder and Michael addition reactions in water using bio-inspired triazacyclophane-based ligands. Eur. J. Org. Chem. 2011, 2011, 1714–1720. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder reaction in total synthesis. Angew. Chem. Int. Ed. Engl. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Takao, K.; Munakata, R.; Tadano, K. Recent advances in natural product synthesis by using intramolecular Diels–Alder reactions. Chem. Rev. 2005, 105, 4779–4807. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Vavsari, V.F. Recent applications of intramolecular Diels–Alder reaction in total synthesis of natural products. RSC Adv. 2015, 5, 50890–50912. [Google Scholar] [CrossRef]
- Lam, H.C.; Pepper, H.P.; Sumby, C.J.; George, J.H. Biomimetic total synthesis of (±)-verrubenzospirolactone. Angew. Chem. Int. Ed. Engl. 2017, 56, 8532–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
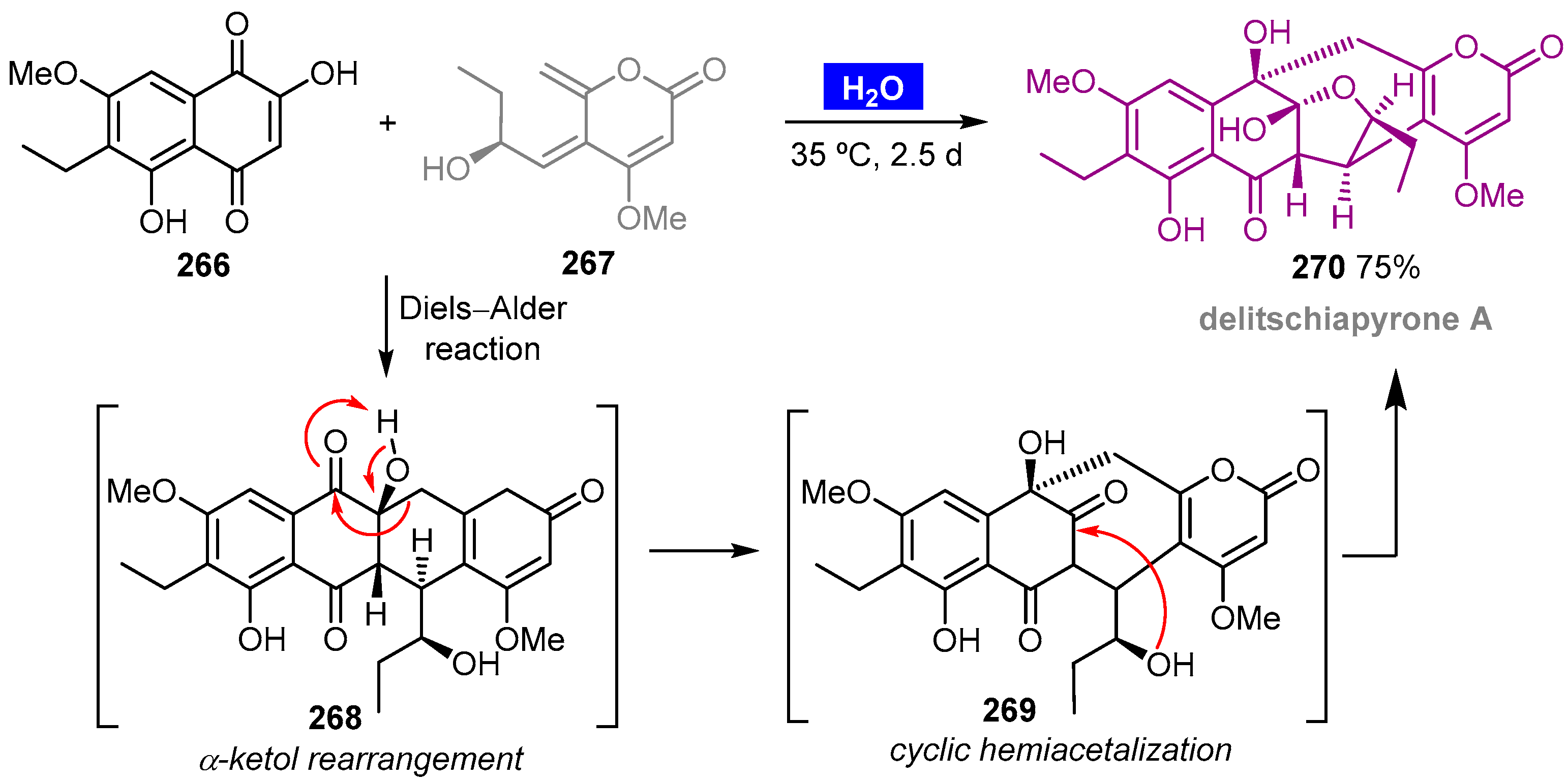
- Kurasawa, K.; Kwon, E.; Kuwahara, S.; Enomoto, M. Bioinspired total synthesis of delitschiapyrone A. Org. Lett. 2018, 20, 4645–4648. [Google Scholar] [CrossRef] [PubMed]












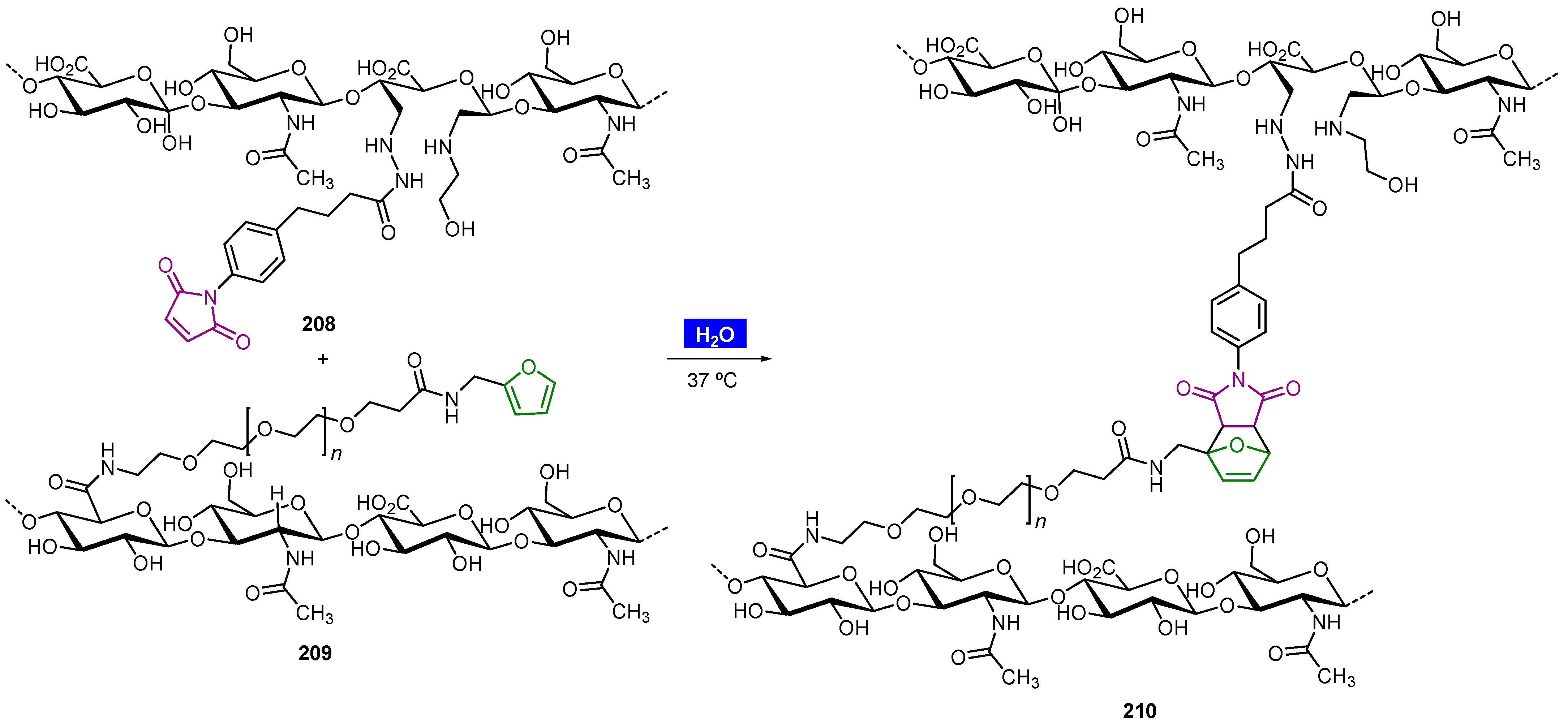


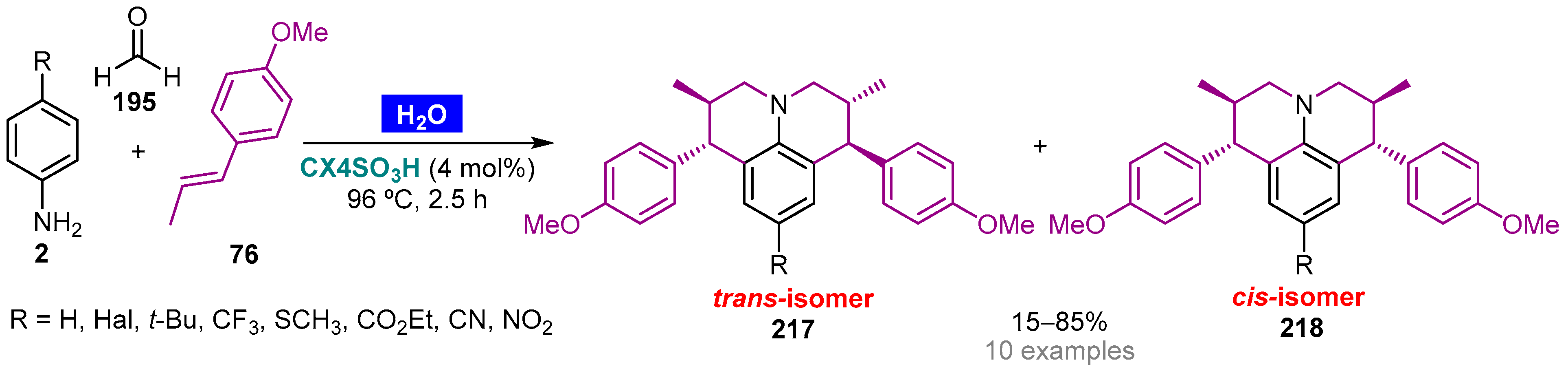
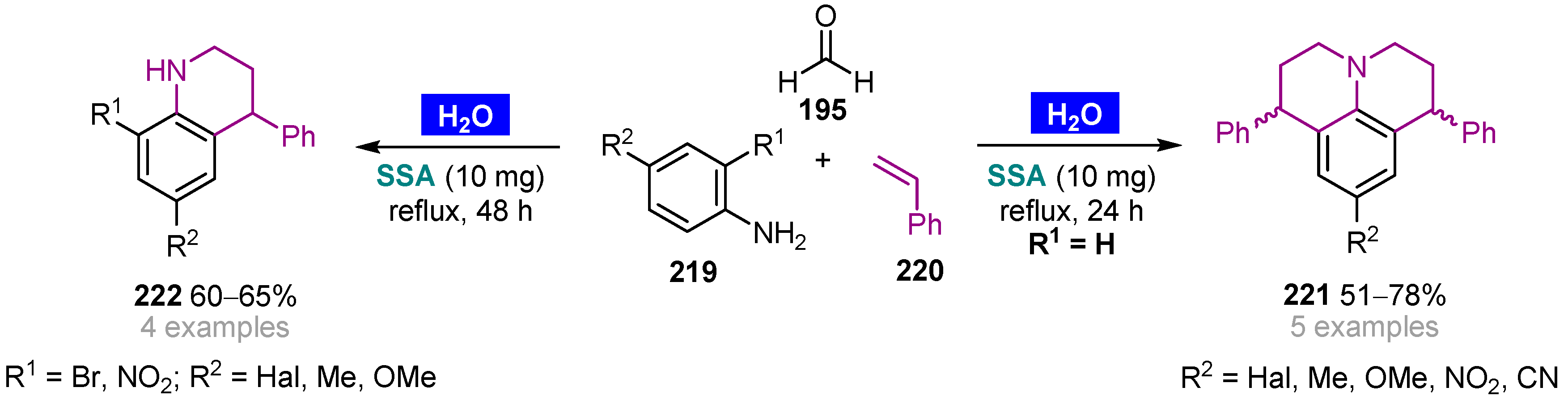
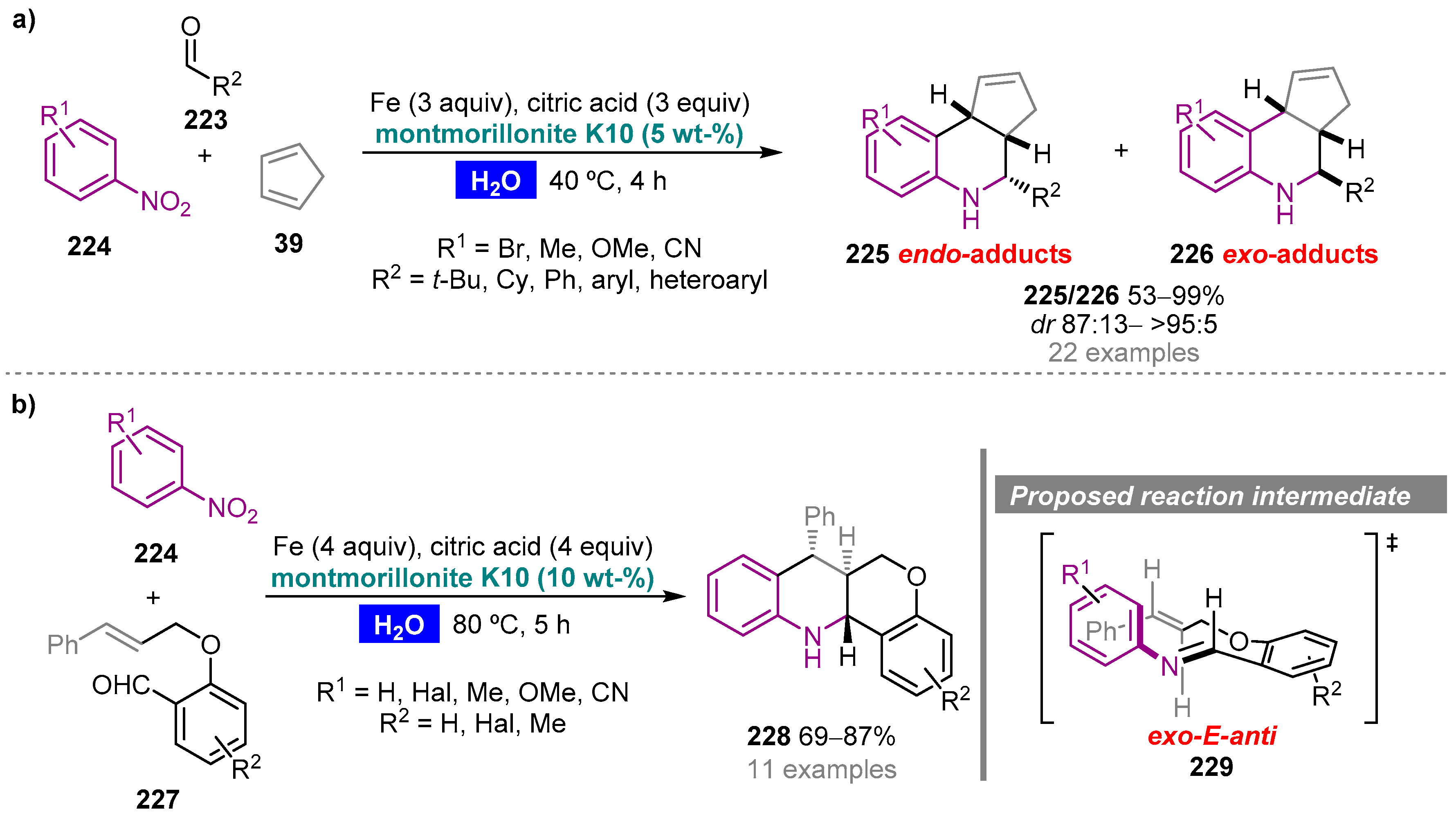
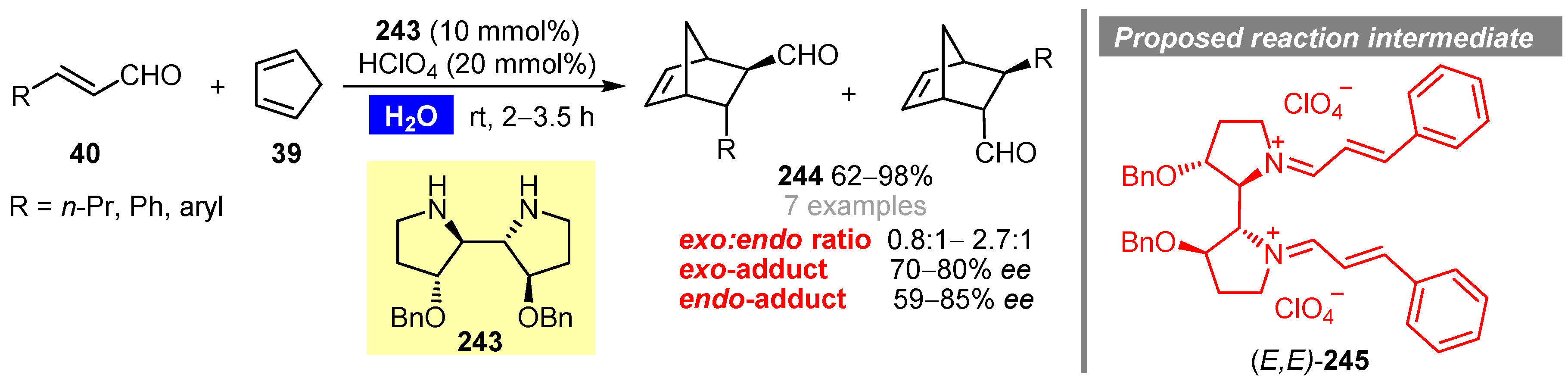
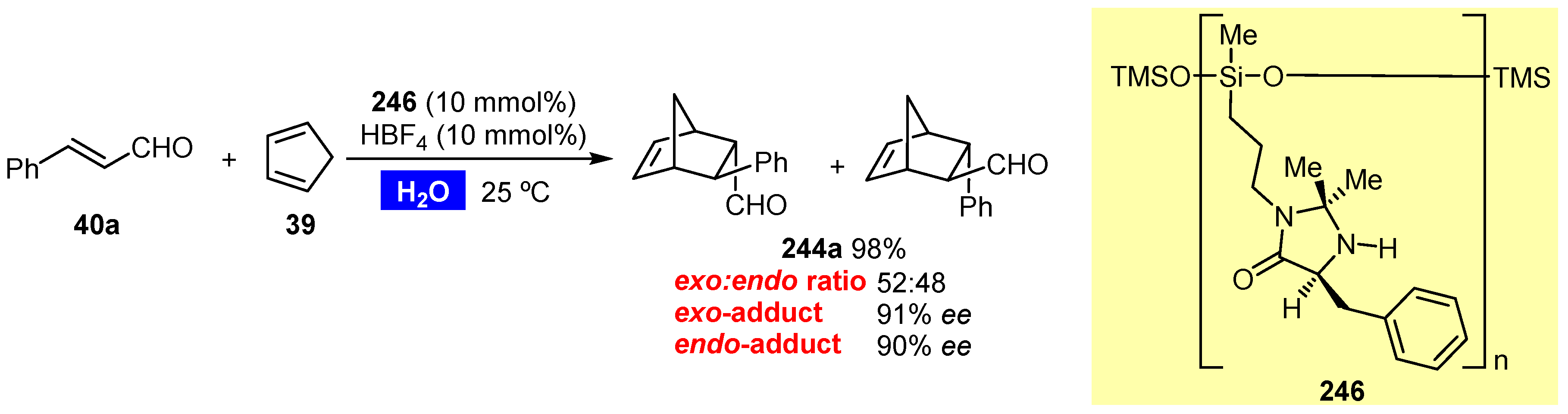


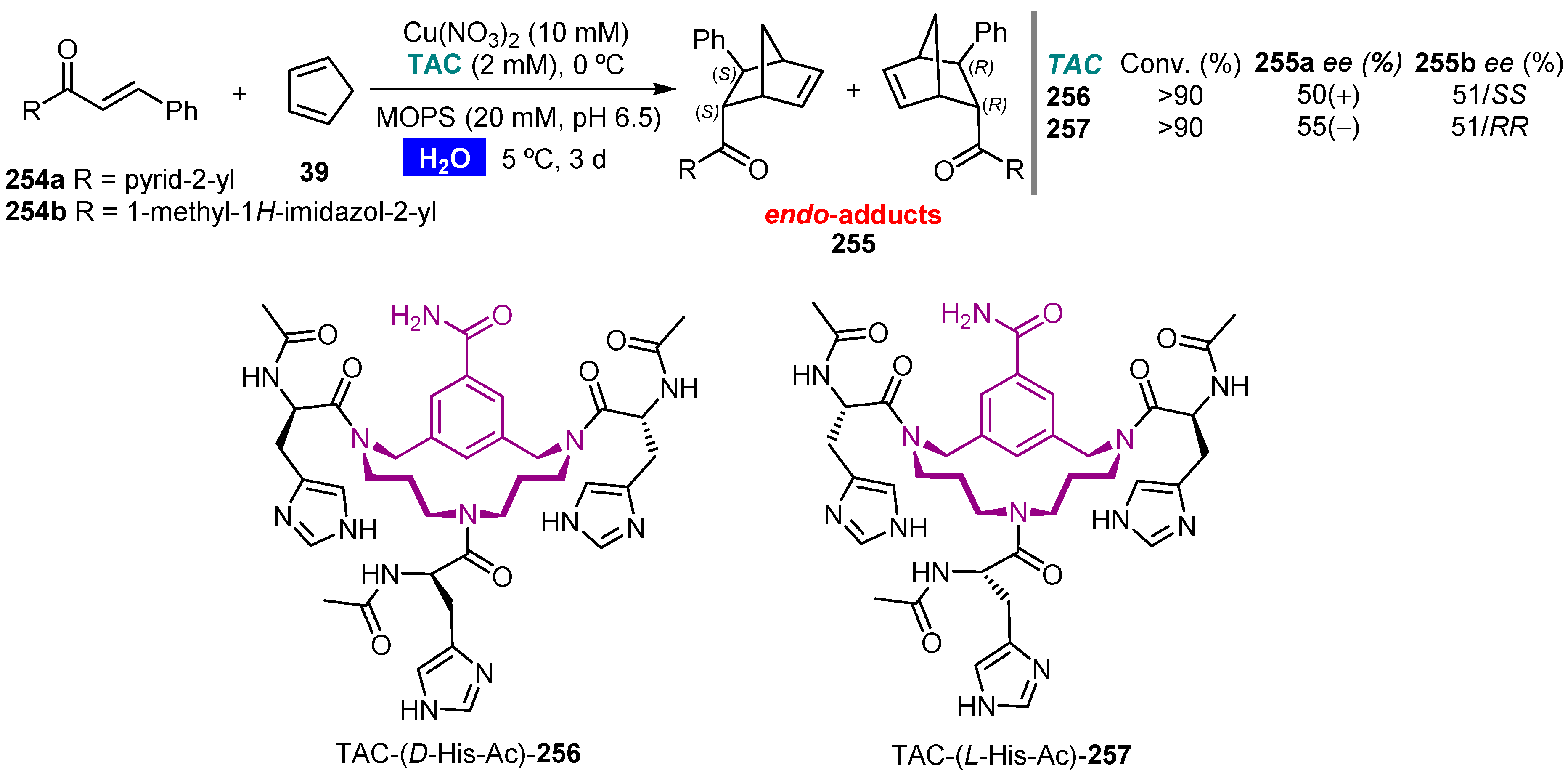
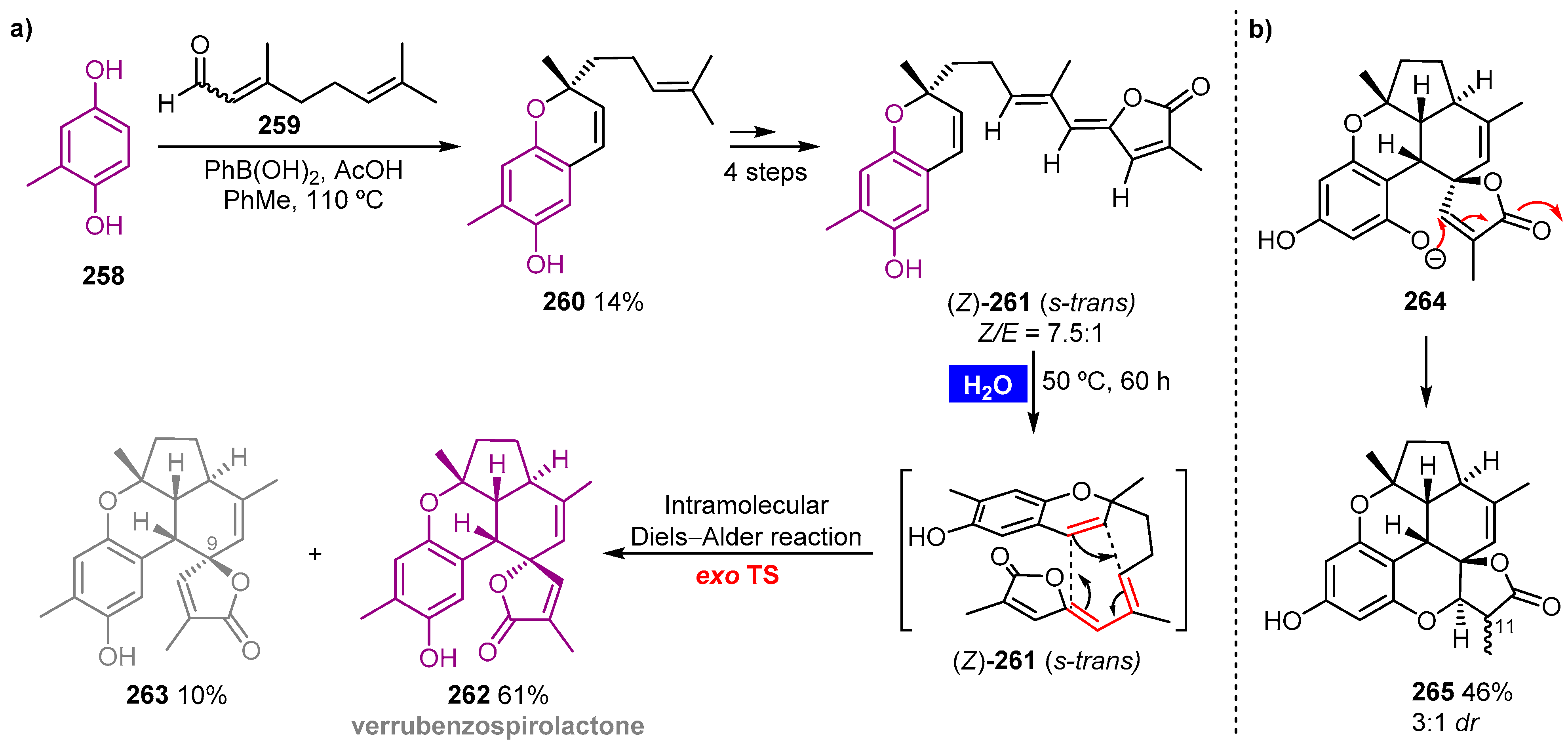
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, M.I.L.; Cardoso, A.L.; Pinho e Melo, T.M.V.D. Diels–Alder Cycloaddition Reactions in Sustainable Media. Molecules 2022, 27, 1304. https://doi.org/10.3390/molecules27041304
Soares MIL, Cardoso AL, Pinho e Melo TMVD. Diels–Alder Cycloaddition Reactions in Sustainable Media. Molecules. 2022; 27(4):1304. https://doi.org/10.3390/molecules27041304
Chicago/Turabian StyleSoares, Maria I. L., Ana L. Cardoso, and Teresa M. V. D. Pinho e Melo. 2022. "Diels–Alder Cycloaddition Reactions in Sustainable Media" Molecules 27, no. 4: 1304. https://doi.org/10.3390/molecules27041304