
Improved Cell-Potent and Selective Peptidomimetic Inhibitors of Protein N-Terminal Methyltransferase 1


Abstract
:1. Introduction
2. Design
3. Synthesis
4. Structure–Activity Relationship
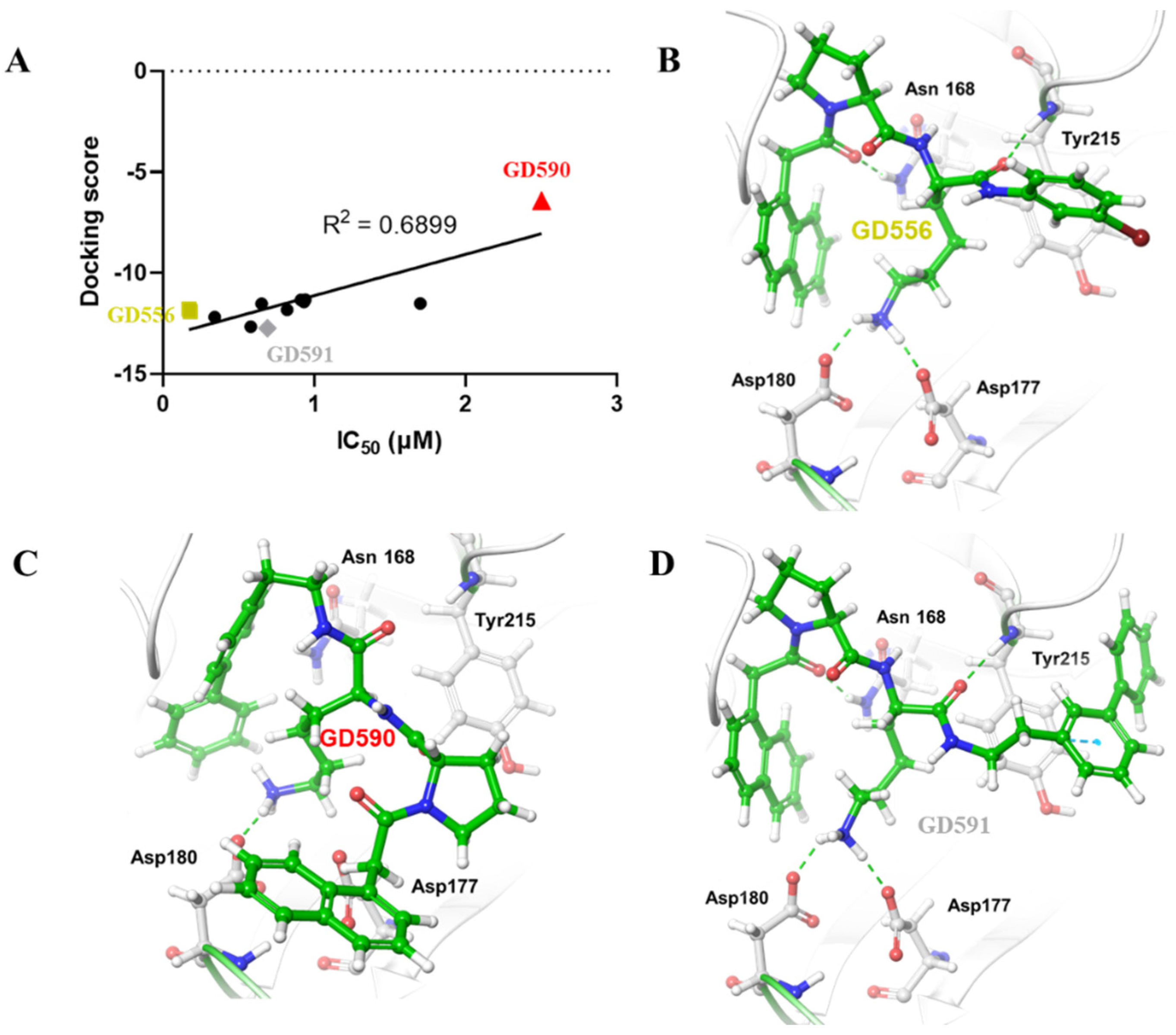
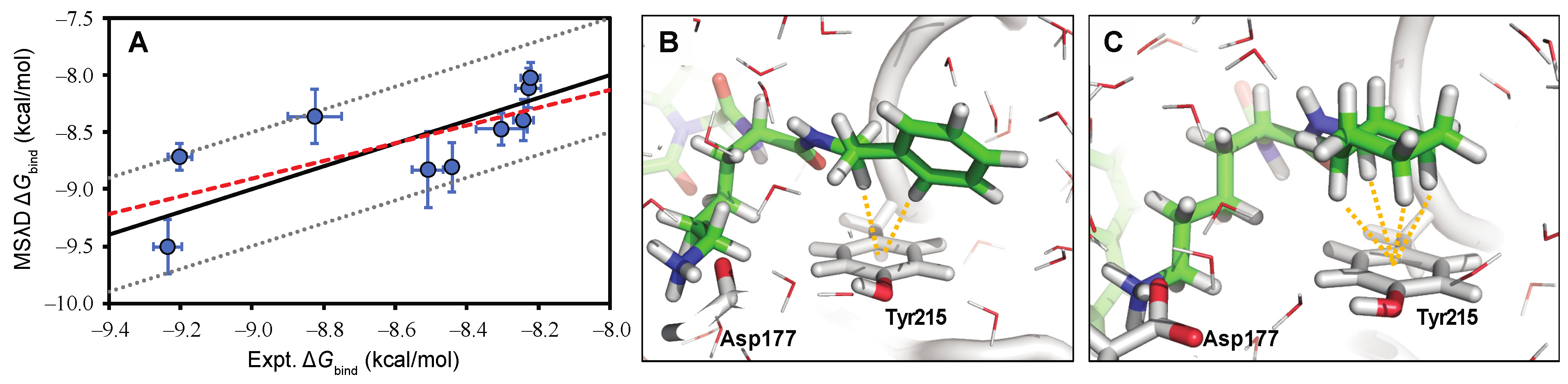
5. Computational Studies
6. Cytotoxicity
7. Inhibition on Cellular N-Terminal Methylation
8. Permeability
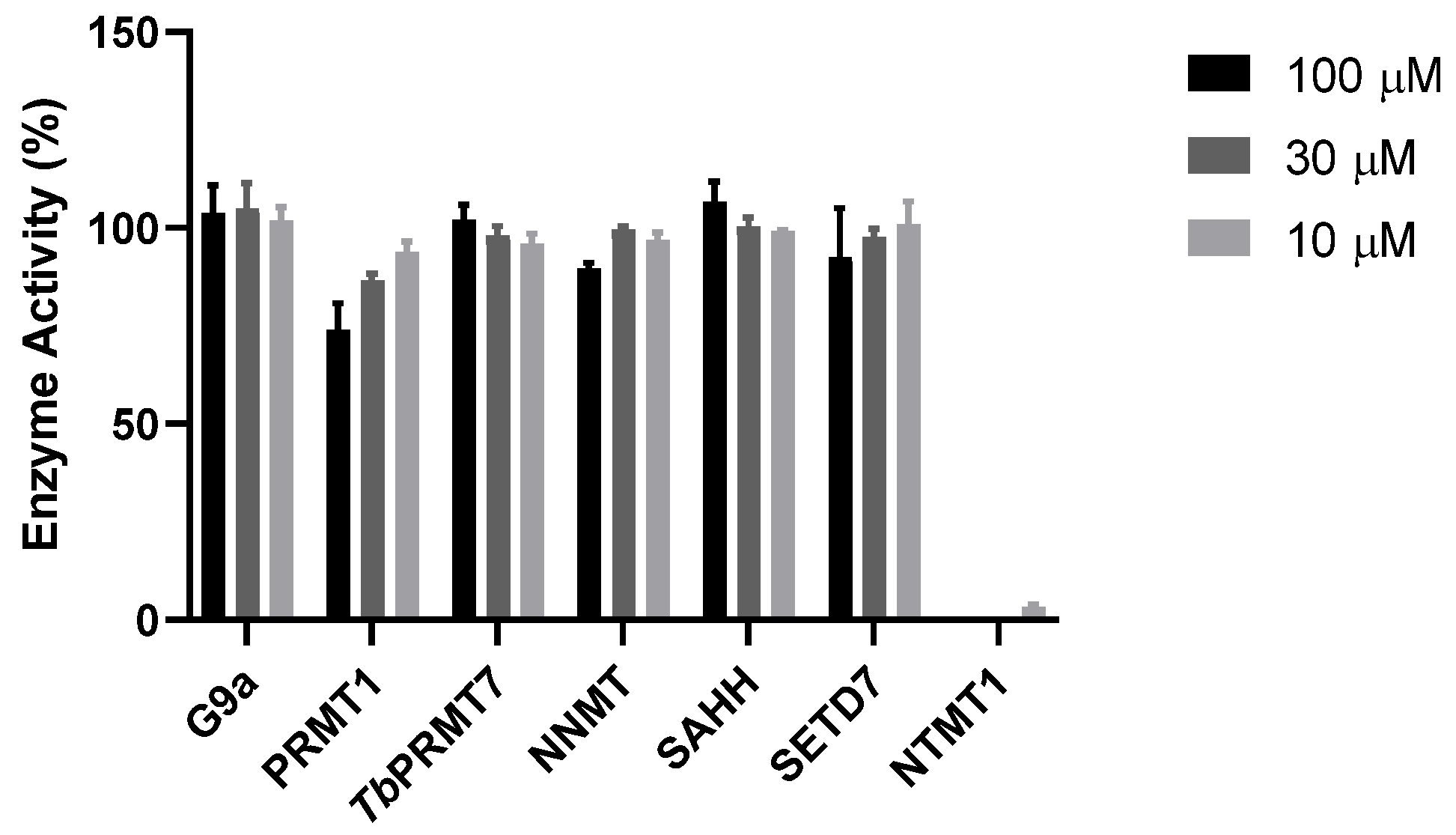
9. Selectivity
10. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schaner Tooley, C.E.; Petkowski, J.J.; Muratore-Schroeder, T.L.; Balsbaugh, J.L.; Shabanowitz, J.; Sabat, M.; Minor, W.; Hunt, D.F.; Macara, I.G. NRMT is an α-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 2010, 466, 1125–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petkowski, J.J.; Bonsignore, L.A.; Tooley, J.G.; Wilkey, D.W.; Merchant, M.L.; Macara, I.G.; Schaner Tooley, C.E. NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem. J. 2013, 456, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathyan, K.M.; Fachinetti, D.; Foltz, D.R. α-amino trimethylation of CENP-A by NRMT is required for full recruitment of the centromere. Nat. Commun. 2017, 8, 14678. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Fu, L.; Wang, Z.; Gan, N.; Dai, X.; Wang, Y. α-N-Methylation of Damaged DNA-Binding Protein 2 (DDB2) and Its Function in Nucleotide Excision Repair. J. Biol. Chem. 2014, 289, 16046–16056. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Rulten, S.L.; You, C.; Caldecott, K.W.; Wang, Y. Identification and Functional Characterizations of N-Terminal α-N-Methylation and Phosphorylation of Serine 461 in Human Poly(ADP-ribose) Polymerase 3. J. Proteome Res. 2015, 14, 2575–2582. [Google Scholar] [CrossRef] [Green Version]
- Jia, K.; Huang, G.; Wu, W.; Shrestha, R.; Wu, B.; Xiong, Y.; Li, P. In vivo methylation of OLA1 revealed by activity-based target profiling of NTMT1. Chem. Sci. 2019, 10, 8094–8099. [Google Scholar] [CrossRef] [Green Version]
- Bade, D.; Cai, Q.; Li, L.; Yu, K.; Dai, X.; Miao, W.; Wang, Y. Modulation of N-terminal methyltransferase 1 by an N(6)-methyladenosine-based epitranscriptomic mechanism. Biochem. Biophys. Res. Commun. 2021, 546, 54–58. [Google Scholar] [CrossRef]
- Huang, R. Chemical Biology of Protein N-Terminal Methyltransferases. Chembiochem 2019, 20, 976–984. [Google Scholar] [CrossRef]
- Grimsley, G.R.; Scholtz, J.M.; Pace, C.N. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009, 18, 247–251. [Google Scholar]
- Chen, T.; Muratore, T.L.; Schaner-Tooley, C.E.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G. N-terminal α-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat. Cell Biol. 2007, 9, 596. [Google Scholar] [CrossRef]
- Bonsignore, L.A.; Butler, J.S.; Klinge, C.M.; Schaner Tooley, C.E. Loss of the N-terminal methyltransferase NRMT1 increases sensitivity to DNA damage and promotes mammary oncogenesis. Oncotarget 2015, 6, 12248–12263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, H.; Chen, C.; Chen, L.; Dai, Y.; Sun, P.H.; Zou, C.; Wang, X. Methyltransferase-like protein 11A promotes migration of cervical cancer cells via up-regulating ELK3. Pharmacol. Res. 2021, 172, 105814. [Google Scholar] [CrossRef] [PubMed]
- Catlin, J.P.; Marziali, L.N.; Rein, B.; Yan, Z.; Feltri, M.L.; Schaner Tooley, C.E. Age-related neurodegeneration and cognitive impairments of NRMT1 knockout mice are preceded by misregulation of RB and abnormal neural stem cell development. Cell Death Dis. 2021, 12, 1014. [Google Scholar] [CrossRef]
- Mackie, B.D.; Chen, D.; Dong, G.; Dong, C.; Parker, H.; Schaner Tooley, C.E.; Noinaj, N.; Min, J.; Huang, R. Selective Peptidomimetic Inhibitors of NTMT1/2: Rational Design, Synthesis, Characterization, and Crystallographic Studies. J. Med. Chem. 2020, 63, 9512–9522. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dong, G.; Deng, Y.; Noinaj, N.; Huang, R. Structure-based Discovery of Cell-Potent Peptidomimetic Inhibitors for Protein N-Terminal Methyltransferase 1. ACS Med. Chem. Lett. 2021, 12, 485–493. [Google Scholar] [CrossRef]
- Zhang, L.; Torgerson, T.R.; Liu, X.Y.; Timmons, S.; Colosia, A.D.; Hawiger, J.; Tam, J.P. Preparation of functionally active cell-permeable peptides by single-step ligation of two peptide modules. Proc. Natl. Acad. Sci. USA 1998, 95, 9184–9189. [Google Scholar] [CrossRef] [Green Version]
- Ben-Dror, S.; Bronshtein, I.; Wiehe, A.; Roder, B.; Senge, M.O.; Ehrenberg, B. On the correlation between hydrophobicity, liposome binding and cellular uptake of porphyrin sensitizers. Photochem. Photobiol. 2006, 82, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Xu, Z. Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Mar. Drugs 2021, 19, 311. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Brooks, C.L., III. λ Dynamics: A New Approach to Free Energy Calculations. J. Chem. Phys. 1996, 105, 2414–2423. [Google Scholar] [CrossRef]
- Knight, J.L.; Brooks, C.L., III. Multisite λ Dynamics for Simulated Structure-Activity Relationship Studies. J. Chem. Theory Comput. 2011, 7, 2728–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keränen, H.; Pérez-Benito, L.; Ciordia, M.; Delgado, F.; Steinbrecher, T.B.; Oehlrich, D.; van Vlijmen, H.W.T.; Trabanco, A.A.; Tresadern, G. Acylguanidine Beta Secretase 1 Inhibitors: A Combined Experimental and Free Energy Perturbation Study. J. Chem. Theory Comput. 2017, 13, 1439–1453. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Severance, D.L. Aromatic-Aromatic Interactions: Free Energy Profiles for the Benzene Dimer in Water, Chloroform, and Liquid Benzene. J. Am. Chem. Soc. 1990, 112, 4768–4774. [Google Scholar] [CrossRef]
- Nishio, M. CH/π hydrogen bonds in crystals. Cryst. Eng. Comm. 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Shields, K.M.; Tooley, J.G.; Petkowski, J.J.; Wilkey, D.W.; Garbett, N.C.; Merchant, M.L.; Cheng, A.; Schaner Tooley, C.E. Select Human Cancer Mutants of NRMT1 Alter Its Catalytic Activity and Decrease N-Terminal Trimethylation. Protein Sci. 2017, 26, 1639–1652. [Google Scholar] [CrossRef] [Green Version]
- Kansy, M.; Senner, F.; Gubernator, K. Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef]
- Avdeef, A.; Strafford, M.; Block, E.; Balogh, M.P.; Chambliss, W.; Khan, I. Drug Absorption in Vitro Model: Filter-Immobilized Artificial Membranes: 2. Studies of the Permeability Properties of Lactones in Piper Methysticum Forst. Eur. J. Pharm. Sci. 2001, 14, 271–280. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| ID | R | IC50 (μM) | cLogP | IC50/IC50 (DC541) |
|---|---|---|---|---|
| DC541 |  | 0.34 ± 0.03 | 0.7 | 1.0 |
| 1a (GD556) | 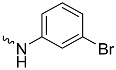 | 0.17 ± 0.008 | 2.9 | 0.50 |
| 1b (GD558) | 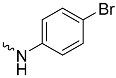 | 0.18 ± 0.007 | 2.9 | 0.53 |
| 1c (GD560) | 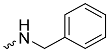 | 0.91 ± 0.03 | 1.7 | 2.7 |
| 1d (GD562) | 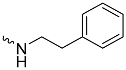 | 0.93 ± 0.04 | 1.9 | 2.7 |
| 1e (GD573) | 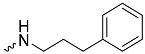 | 0.65 ± 0.01 | 2.3 | 1.9 |
| 1f (GD564) |  | 0.94 ± 0.03 | 2.4 | 2.8 |
| 1g (GD566) | 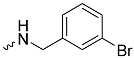 | 0.58 ± 0.03 | 2.5 | 1.7 |
| 1h (GD568) | 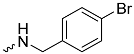 | 0.82 ± 0.07 | 2.5 | 2.4 |
| 1i (GD589) |  | 1.7 ± 0.07 | 2.3 | 5.0 |
| 1j (GD590) | 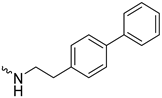 | 2.5 ± 0.09 | 3.8 | 7.4 |
| 1k (GD591) | 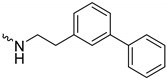 | 0.69 ± 0.08 | 3.8 | 2.0 |
| ID | GI50 (μM) | Cellular IC50 (μM) | Permeability (10−6 cm/s) |
|---|---|---|---|
| 1c (GD560) | >300 | <300 | 0.24 ± 0.06 |
| 1d (GD562) | >300 | ~50 | 0.26 ± 0.06 |
| 1e (GD573) | >300 | ~100 | 0.17 ± 0.08 |
| 1f (GD564) | >300 | ~100 | 0.47 ± 0.07 |
| 1g (GD566) | 128 ± 20 | <300 | 0.6 ± 0.1 |
| 1h (GD568) | 135 ± 23 | <300 | 0.9 ± 0.2 |
| 1j (GD590) | N.D | N.D. | 0.9 ± 0.3 |
| 1k (GD591) | 65 ± 7 | N.D. | 1.3 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, G.; Iyamu, I.D.; Vilseck, J.Z.; Chen, D.; Huang, R. Improved Cell-Potent and Selective Peptidomimetic Inhibitors of Protein N-Terminal Methyltransferase 1. Molecules 2022, 27, 1381. https://doi.org/10.3390/molecules27041381
Dong G, Iyamu ID, Vilseck JZ, Chen D, Huang R. Improved Cell-Potent and Selective Peptidomimetic Inhibitors of Protein N-Terminal Methyltransferase 1. Molecules. 2022; 27(4):1381. https://doi.org/10.3390/molecules27041381
Chicago/Turabian StyleDong, Guangping, Iredia D. Iyamu, Jonah Z. Vilseck, Dongxing Chen, and Rong Huang. 2022. "Improved Cell-Potent and Selective Peptidomimetic Inhibitors of Protein N-Terminal Methyltransferase 1" Molecules 27, no. 4: 1381. https://doi.org/10.3390/molecules27041381
APA StyleDong, G., Iyamu, I. D., Vilseck, J. Z., Chen, D., & Huang, R. (2022). Improved Cell-Potent and Selective Peptidomimetic Inhibitors of Protein N-Terminal Methyltransferase 1. Molecules, 27(4), 1381. https://doi.org/10.3390/molecules27041381