
Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
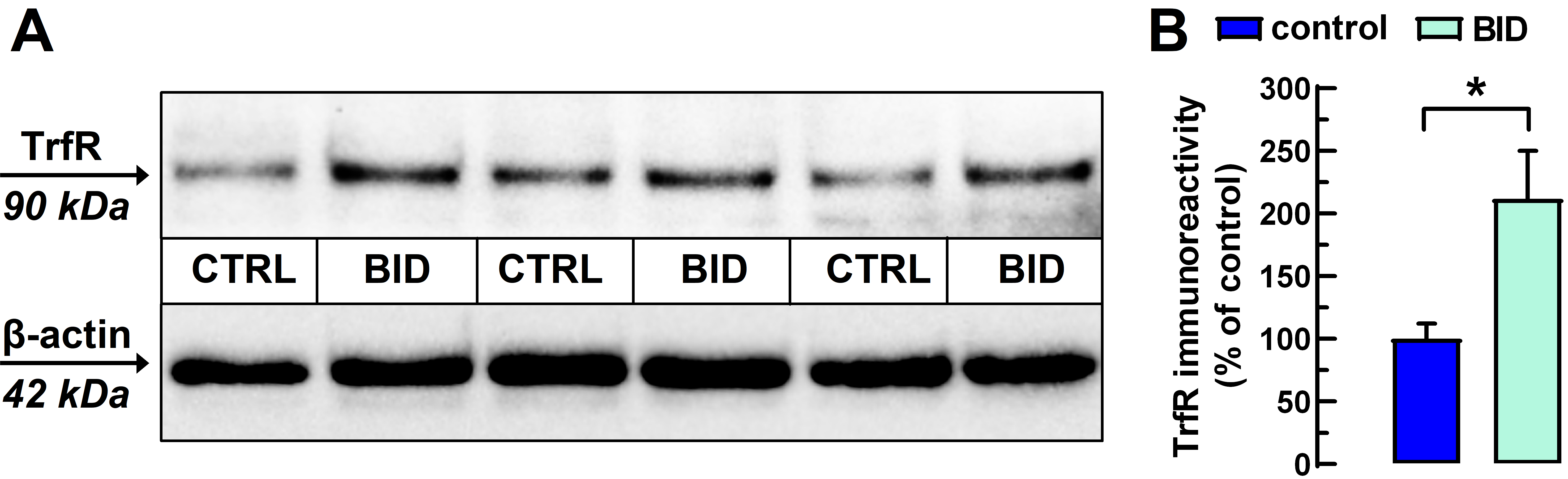
2.1. Diet-Induced Anemic Phenotype and Bid
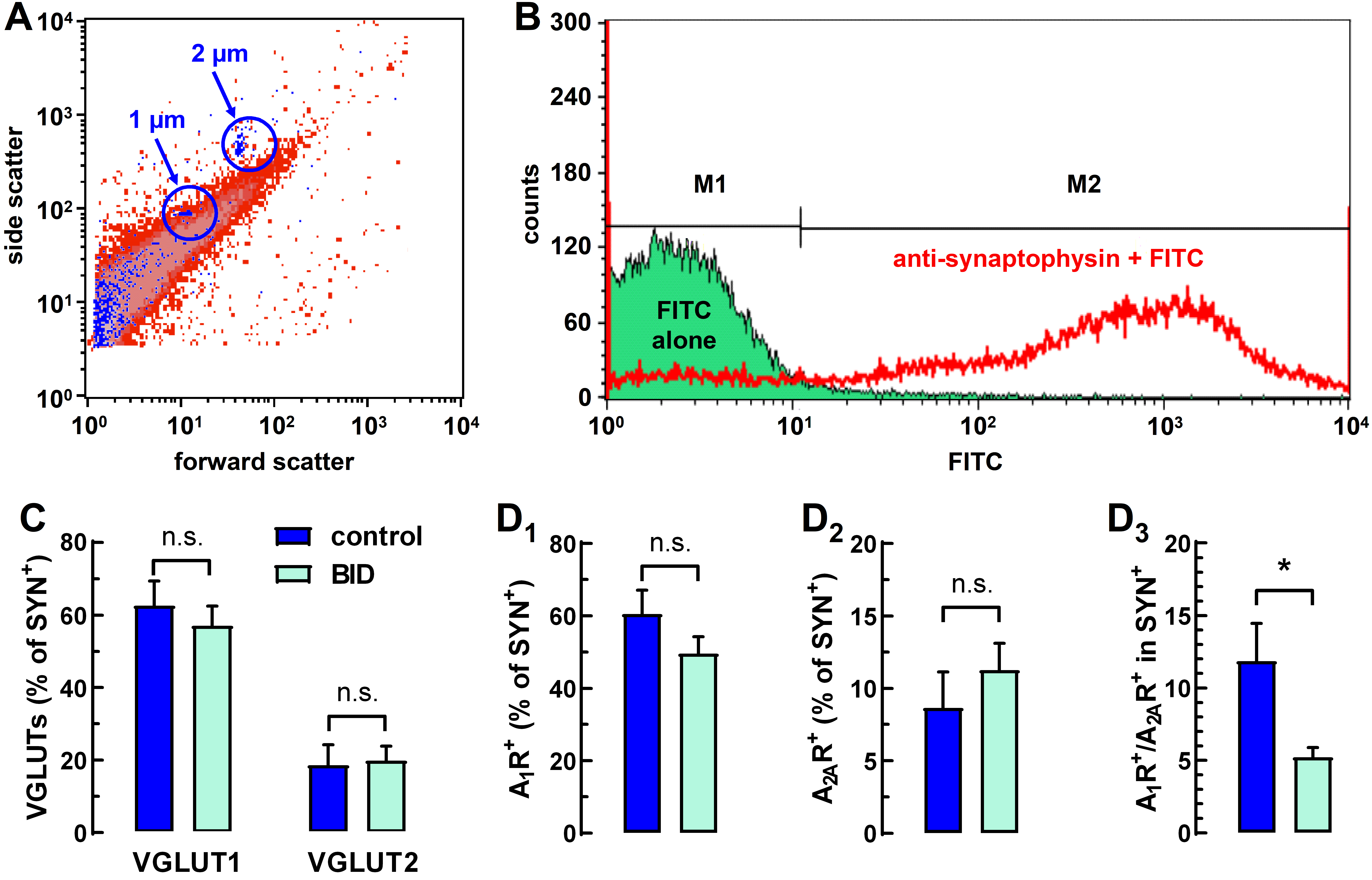
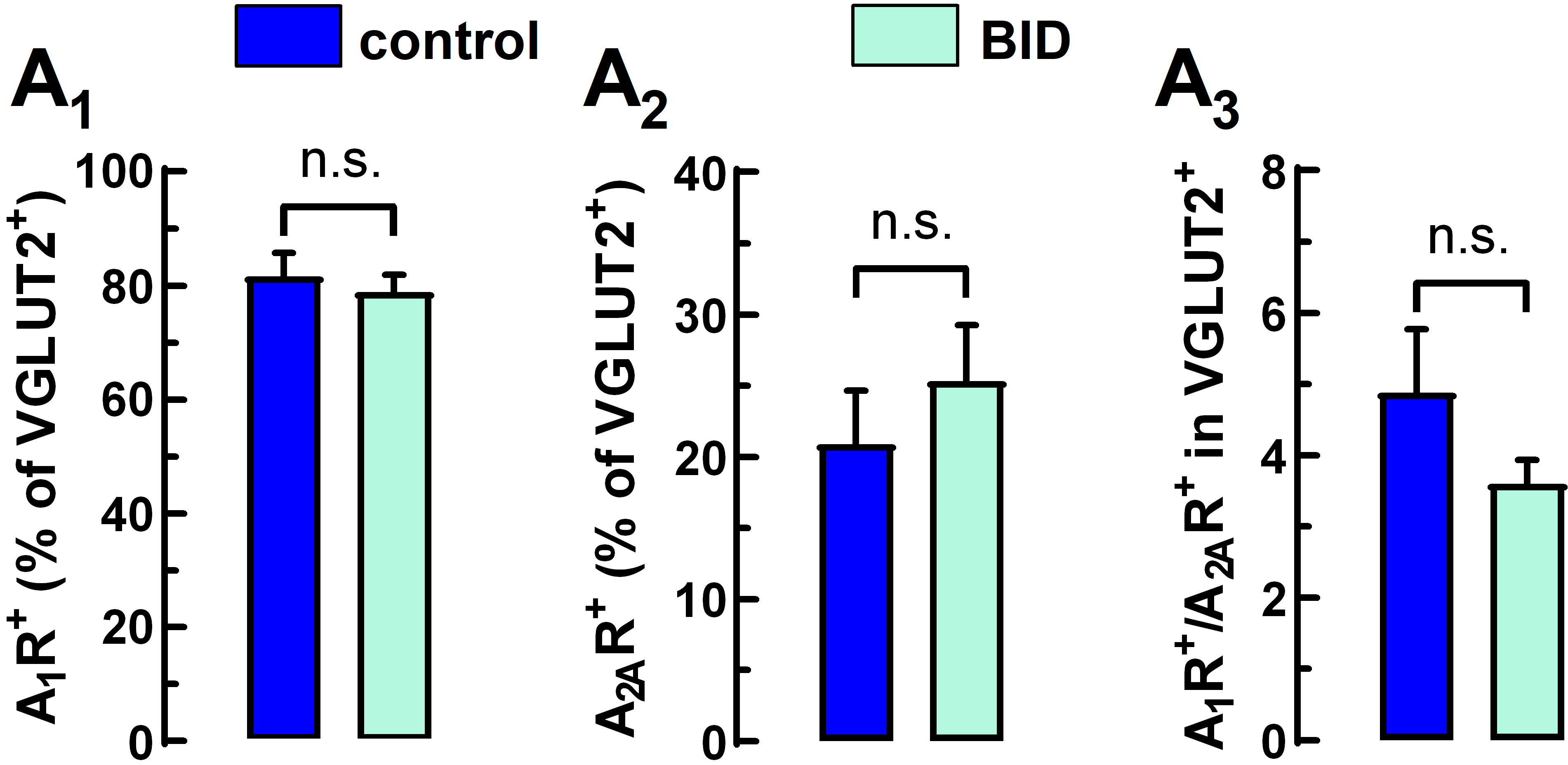
2.2. Flow-Synaptometric Analysis of Striatal Nerve Terminals
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Determination of Peripheral Iron Deficiency and Bid
4.3. Purified Synaptosomes
4.3.1. Preparation of S1 Fraction
4.3.2. Discontinuous Percoll Gradient
4.3.3. Immunolabeling and Flow Synaptometry Analysis
4.3.4. Detection of Synaptosomes and Data Analysis
4.4. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Allen, R.P.; Picchietti, D.L.; Garcia-Borreguero, D.; Ondo, W.G.; Walters, A.S.; Winkelman, J.W.; Zucconi, M.; Ferri, R.; Trenkwalder, C.; Lee, H.B. Restless Legs Syndrome Study Group. Restless legs syndrome/Willis–Ekbom disease diagnostic criteria: Updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria—History, rationale, description, and significance. Sleep Med. 2014, 15, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Manconi, M.; Garcia-Borreguero, D.; Schormair, B.; Videnovic, A.; Berger, K.; Ferri, R.; Dauvilliers, Y. Restless legs syndrome. Nat. Rev. Dis. Primers 2021, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; García-Borreguero, D.; Allen, R.P.; Earley, C.J. New Insights into the Neurobiology of Restless Legs Syndrome. Neuroscientist 2018, 25, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Earley, C.J.; Connor, J.; Garcia-Borreguero, D.; Jenner, P.; Winkelman, J.; Zee, P.C.; Allen, R. Altered Brain iron homeostasis and dopaminergic function in Restless Legs Syndrome (Willis–Ekbom Disease). Sleep Med. 2014, 15, 1288–1301. [Google Scholar] [CrossRef]
- Ferré, S.; Quiroz, C.; Guitart, X.; Rea, W.; Seyedian, A.; Moreno, E.; Casadó-Anguera, V.; Díaz-Ríos, M.; Casadó, V.; Clemens, S.; et al. Pivotal Role of Adenosine Neurotransmission in Restless Legs Syndrome. Front. Neurosci. 2018, 11, 722. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Borreguero, D.; Guitart, X.; Garcia Malo, C.; Cano-Pumarega, I.; Granizo, J.J.; Ferré, S. Treatment of restless legs syn-drome/Willis-Ekbom disease with the non-selective ENT1/ENT2 inhibitor dipyridamole: Testing the adenosine hypothesis. Sleep Med. 2018, 45, 94–97. [Google Scholar] [CrossRef]
- Garcia-Borreguero, D.; Garcia-Malo, C.; Granizo, J.J.; Ferré, S. A Randomized, Placebo-Controlled Crossover Study with Dipyridamole for Restless Legs Syndrome. Mov. Disord. 2021, 36, 2387–2392. [Google Scholar] [CrossRef]
- Quiroz, C.; Pearson, V.; Gulyani, S.; Allen, R.; Earley, C.; Ferré, S. Up-regulation of striatal adenosine A2A receptors with iron deficiency in rats: Effects on locomotion and cortico-striatal neurotransmission. Exp. Neurol. 2010, 224, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, C.; Gulyani, S.; Ruiqian, W.; Bonaventura, J.; Cutler, R.; Pearson, V.; Allen, R.P.; Earley, C.J.; Mattson, M.P.; Ferré, S. Adenosine receptors as markers of brain iron deficiency: Implications for Restless Legs Syndrome. Neuropharmacology 2016, 111, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Dunwiddie, T.V.; Masino, S.A. The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Fuxe, K.; Fredholm, B.B.; Morelli, M.; Popoli, P. Adenosine-dopamine receptor-receptor interactions as an integra-tive mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar] [CrossRef]
- Borycz, J.; Pereira, M.F.; Melani, A.; Rodrigues, R.J.; Köfalvi, A.; Panlilio, L.; Pedata, F.; Goldberg, S.R.; Cunha, R.A.; Ferré, S. Differential glutamate-dependent and glutamate-independent adenosine A1 receptor-mediated modulation of dopamine re-lease in different striatal compartments. J. Neurochem. 2007, 101, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.; Basheer, R.; McKenna, J.; Strecker, R.E.; McCarley, R. Control of Sleep and Wakefulness. Physiol. Rev. 2012, 92, 1087–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yepes, G.; Guitart, X.; Rea, W.; Newman, A.H.; Allen, R.P.; Earley, C.J.; Quiroz, C.; Ferré, S. Targeting hypersensitive cor-ticostriatal terminals in restless legs syndrome. Ann. Neurol. 2017, 82, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Silber, M.H.; Buchfuhrer, M.J.; Earley, C.J.; Koo, B.B.; Manconi, M.; Winkelman, J.W. The management of Restless Legs Syndrome: An updated algorithm. Mayo Clin. Proc. 2021, 96, 1921–1937. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, J.; Quiroz, C.; Cai, N.S.; Rubinstein, M.; Tanda, G.; Ferré, S. Key role of the dopamine D4 receptor in the mod-ulation of corticostriatal glutamatergic neurotransmission. Sci. Adv. 2017, 3, e1601631. [Google Scholar] [CrossRef] [Green Version]
- Ciruela, F.; Casadó, V.; Rodrigues, R.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.; Cunha, R.; Kull, B.; Fredholm, B.; Ribeiro, J. Adenosine A2A receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1 receptor inhibition. Neuroscience 2002, 112, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Köfalvi, A.; Moreno, E.; Cordomí, A.; Cai, N.S.; Fernández-Dueñas, V.; Ferreira, S.G.; Guixà-González, R.; Sánchez-Soto, M.; Yano, H.; Casadó-Anguera, V.; et al. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biol. 2020, 18, 9. [Google Scholar] [CrossRef]
- Ferré, S.; Quiroz, C.; Rea, W.; Guitart, X.; García-Borreguero, D. Adenosine mechanisms and hypersensitive corticostriatal terminals in restless legs syndrome. Rationale for the use of inhibitors of adenosine transport. Adv. Pharmacol. 2019, 84, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Lok, C.; Loh, T. Regulation of transferrin function and expression: Review and update. Biol. Signals Recept. 1998, 7, 157–178. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Day, J.R.; Connor, J.R.; Beard, J.L. Gene expression of transferrin and transferrin receptor in brains of control vs. iron-deficient rats. Nutr. Neurosci. 2003, 6, 1–10. [Google Scholar]
- Gulyani, S.; Earley, C.J.; Camandola, S.; Maudsley, S.; Ferré, S.; Mughal, M.R.; Martin, B.; Cheng, A.; Gleichmann, M.; Jones, B.C.; et al. Diminished iron concentrations increase adenosine A2A receptor levels in mouse striatum and cultured human neuroblastoma cells. Exp. Neurol. 2009, 215, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.G.; Gonçalves, F.Q.; Marques, J.M.; Tomé, R.; Rodrigues, R.J.; Nunes-Correia, I.; Ledent, C.; Harkany, T.; Venance, L.; A Cunha, R.; et al. Presynaptic adenosine A2A receptors dampen cannabinoid CB1 receptor-mediated inhibition of corticostriatal glutamatergic transmission. J. Cereb. Blood Flow Metab. 2015, 172, 1074–1086. [Google Scholar] [CrossRef] [Green Version]
- Fujiyama, F.; Kuramoto, E.; Okamoto, K.; Hioki, H.; Furuta, T.; Zhou, L.; Nomura, S.; Kaneko, T. Presynaptic localization of an AMPA-type glutamate receptor in corticostriatal and thalamostriatal axon terminals. Eur. J. Neurosci. 2004, 20, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, F.; Unzai, T.; Nakamura, K.; Nomura, S.; Kaneko, T. Difference in organization of corticostriatal and thalamostri-atal synapses between patch and matrix compartments of rat neostriatum. Eur. J. Neurosci. 2006, 24, 2813–2824. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.V.; Smith, Y. Differential Localization of Vesicular Glutamate Transporters 1 and 2 in the Rat Striatum. Neuroscience 2006, 601–610. [Google Scholar] [CrossRef]
- Navarro, G.; Cordomí, A.; Casadó-Anguera, V.; Moreno, E.; Cai, N.-S.; Cortés, A.; Canela, E.I.; Dessauer, C.W.; Casadó, V.; Pardo, L.; et al. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Ferré, S.; Bonaventura, J.; Zhu, W.; Hatcher-Solis, C.; Taura, J.; Quiroz, C.; Cai, N.-S.; Moreno, E.; Casadó-Anguera, V.; Kravitz, A.V.; et al. Essential Control of the Function of the Striatopallidal Neuron by Pre-coupled Complexes of Adenosine A2A-Dopamine D2 Receptor Heterotetramers and Adenylyl Cyclase. Front. Pharmacol. 2018, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Popoli, P.; Betto, P.; Reggio, R.; Ricciarello, G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur. J. Pharmacol. 1995, 287, 215–217. [Google Scholar] [CrossRef]
- Quarta, D.; Borycz, J.; Solinas, M.; Patkar, K.; Hockemeyer, J.; Ciruela, F.; Lluis, C.; Franco, R.; Woods, A.S.; Goldberg, S.R.; et al. Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-d-aspartate receptor stimulation. J. Neurochem. 2004, 91, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.J.; Alfaro, T.M.; Rebola, N.; Oliveira, C.R.; Cunha, R.A. Co-localization and functional interaction between adenosine A2A and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J. Neurochem. 2004, 92, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.Y.; Canas, P.M.; Garcia-Sanz, P.; Lan, J.Q.; Boison, D.; Moratalla, R.; Cunha, R.A.; Chen, J.F. Adenosine A2A receptors in striatal glutamatergic terminals and GABAergic neurons oppositely modulate psychostimulant action and DARPP-32 phosphorylation. PLoS ONE 2013, 8, e80902. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Carmo, M.; Gonçalves, F.Q.; Canas, P.M.; Oses, J.P.; Fernandes, F.D.; Duarte, F.V.; Palmeira, C.M.; Tomé, A.R.; Agostinho, P.; Andrade, G.M.; et al. Enhanced ATP release and CD73-mediated adenosine formation sustain adenosine A2A receptor over-activation in a rat model of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 3666–3680. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Augusto, E.; Gonçalves, F.Q.; Real, J.E.; Silva, H.B.; Pochmann, D.; Silva, T.S.; Matos, M.; Gonçalves, N.; Tomé, Â.R.; Chen, J.F.; et al. Increased ATP release and CD73-mediated adenosine A2A receptor activation mediate convulsion-associated neuronal damage and hippocampal dysfunction. Neurobiol. Dis. 2021, 157, 105441. [Google Scholar] [CrossRef]
- Augusto, E.; Matos, M.; Sévigny, J.; El-Tayeb, A.; Bynoe, M.S.; Müller, C.E.; Cunha, R.A.; Chen, J.-F. Ecto-5’-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [Green Version]
- Orrù, M.; Bakešová, J.; Brugarolas, M.; Quiroz, C.; Beaumont, V.; Goldberg, S.R.; Lluis, C.; Cortés, A.; Franco, R.; Casadó, V.; et al. Striatal Pre- and Postsynaptic Profile of Adenosine A2A Receptor Antagonists. PLoS ONE 2011, 6, e16088. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, C.; Orrù, M.; Rea, W.; Ciudad-Roberts, A.; Yepes, G.; Britt, J.P.; Ferré, S. Local Control of Extracellular Dopamine Levels in the Medial Nucleus Accumbens by a Glutamatergic Projection from the Infralimbic Cortex. J. Neurosci. 2016, 36, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, K.A.; Tehan, B.; Lebon, G.; Tate, C.G.; Weir, M.; Marshall, F.H.; Langmead, C. Pharmacology and Structure of Isolated Conformations of the Adenosine A2A Receptor Define Ligand Efficacy. Mol. Pharmacol. 2013, 83, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tergau, F.; Wischer, S.; Paulus, W. Motor system excitability in patients with restless legs syndrome. Neurology 1999, 52, 1060. [Google Scholar] [CrossRef] [PubMed]
- Scalise, A.; Cadore, I.P.; Gigli, G.L. Motor cortex excitability in restless legs syndrome. Sleep Med. 2004, 5, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Cantone, M.; Lanuzza, B.; Pennisi, M.; Bella, R.; Pennisi, G.; Ferri, R. Distinctive patterns of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome, restless legs syndrome, insomnia, and sleep deprivation. Sleep Med. Rev. 2015, 19, 39–50. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.; Ribeiro, J. Increase in the Number, G Protein Coupling, and Efficiency of Facilitatory Adenosine A2A Receptors in the Limbic Cortex, but not Striatum, of Aged Rats. J. Neurochem. 2002, 73, 1733–1738. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Cunha, R.A.; de Mendonça, A.; Ribeiro, J.A. Modification of adenosine modulation of synaptic transmission in the hippocampus of aged rats. Br. J. Pharmacol. 2000, 131, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Sebastião, A.M.; de Mendonca, A.; Oliveira, C.R.; Ribeiro, J.A.; Cunha, R.A. Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J. Neurophysiol. 2003, 90, 1295–1303. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Car-valho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef]
- Dunkley, P.R.; E Jarvie, P.; Robinson, P.J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat. Protoc. 2008, 3, 1718–1728. [Google Scholar] [CrossRef]
- Gylys, K.H.; Fein, J.A.; Cole, G.M. Quantitative characterization of crude synaptosomal fraction (P-2) components by flow cytometry. J. Neurosci. Res. 2000, 61, 186–192. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Group | Mean | S.E.M. | p |
|---|---|---|---|---|
| Body Weight (g) | control | 296.4 | 8.47 | 0.0002 |
| BID | 248.5 | 6.62 | ||
| Erythrocytes (1012/L) | control | 6.68 | 0.08 | <0.0001 |
| BID | 2.34 | 0.20 | ||
| Hemoglobin (g/dL) | control | 14.2 | 0.23 | <0.0001 |
| BID | 5.20 | 0.31 | ||
| Hematocrit (L/L) | control | 0.44 | 0.01 | <0.0001 |
| BID | 0.12 | 0.01 | ||
| Mean Corpuscular Volume (fL) | control | 65.1 | 0.54 | 0.0029 |
| BID | 57.3 | 2.76 | ||
| Mean Globular Haemoglobin (pg) | control | 21.1 | 0.24 | 0.287 |
| BID | 26.4 | 2.83 | ||
| Mean Corpuscolar Haemoglobin (g/dL) | control | 32.4 | 0.21 | 0.0093 |
| BID | 44.9 | 3.16 | ||
| Red Cell Distribution Width (%) | control | 12.1 | 0.14 | <0.0001 |
| BID | 23.3 | 2.36 | ||
| Leukocytes | control | 9.28 | 1.06 | 0.0717 |
| BID | 6.43 | 0.97 | ||
| Segmented Neutrophils | control | 1.21 | 0.16 | 0.8642 |
| BID | 1.09 | 0.27 | ||
| Eosinophils | control | 0.08 | 0.05 | 0.4814 |
| BID | 0.054 | 0.03 | ||
| Lymphocytes (109/L) | control | 7.51 | 0.81 | 0.0317 |
| BID | 4.99 | 0.66 | ||
| Monocytes (109/L) | control | 0.45 | 0.11 | 0.1782 |
| BID | 0.29 | 0.09 | ||
| Thrombocytes (109/L) | control | 777.0 | 26.6 | 0.0215 |
| BID | 1247.6 | 131.1 | ||
| Serum Iron Concentration (μmol/L) | control | 44.1 | 2.78 | <0.0001 |
| BID | 4.39 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, M.S.; Ferreira, S.G.; Quiroz, C.; Earley, C.J.; García-Borreguero, D.; Cunha, R.A.; Ciruela, F.; Köfalvi, A.; Ferré, S. Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome. Molecules 2022, 27, 1489. https://doi.org/10.3390/molecules27051489
Rodrigues MS, Ferreira SG, Quiroz C, Earley CJ, García-Borreguero D, Cunha RA, Ciruela F, Köfalvi A, Ferré S. Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome. Molecules. 2022; 27(5):1489. https://doi.org/10.3390/molecules27051489
Chicago/Turabian StyleRodrigues, Matilde S., Samira G. Ferreira, César Quiroz, Christopher J. Earley, Diego García-Borreguero, Rodrigo A. Cunha, Francisco Ciruela, Attila Köfalvi, and Sergi Ferré. 2022. "Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome" Molecules 27, no. 5: 1489. https://doi.org/10.3390/molecules27051489
APA StyleRodrigues, M. S., Ferreira, S. G., Quiroz, C., Earley, C. J., García-Borreguero, D., Cunha, R. A., Ciruela, F., Köfalvi, A., & Ferré, S. (2022). Brain Iron Deficiency Changes the Stoichiometry of Adenosine Receptor Subtypes in Cortico-Striatal Terminals: Implications for Restless Legs Syndrome. Molecules, 27(5), 1489. https://doi.org/10.3390/molecules27051489