
Synthesis and Structure–Activity Relationship of Thioacetamide-Triazoles against Escherichia coli

 ,
,  and
and
Abstract
:1. Introduction
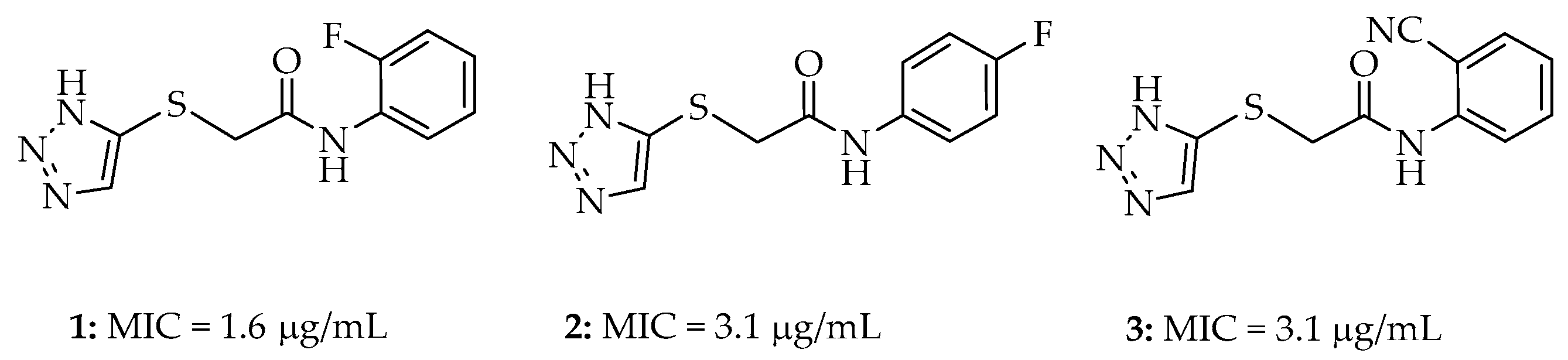
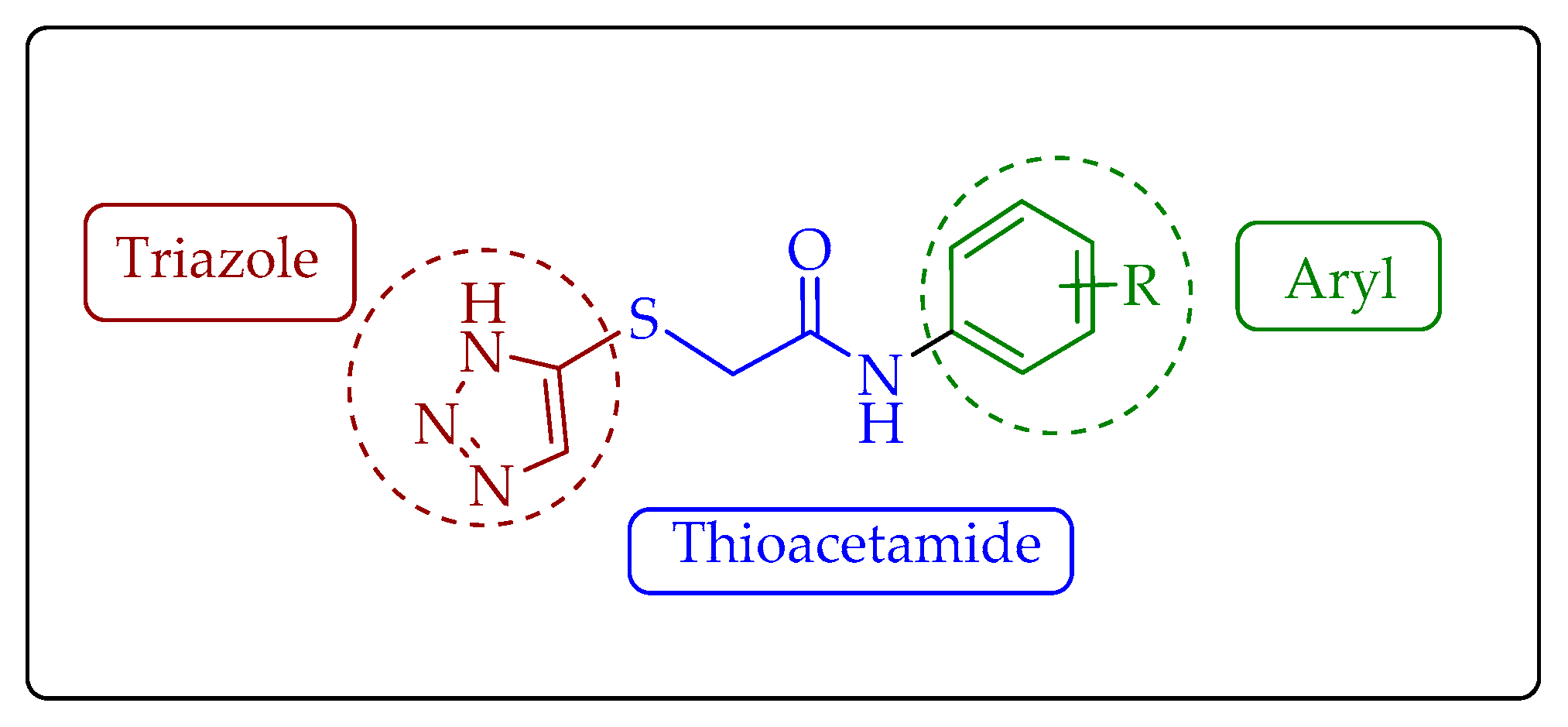
2. Results
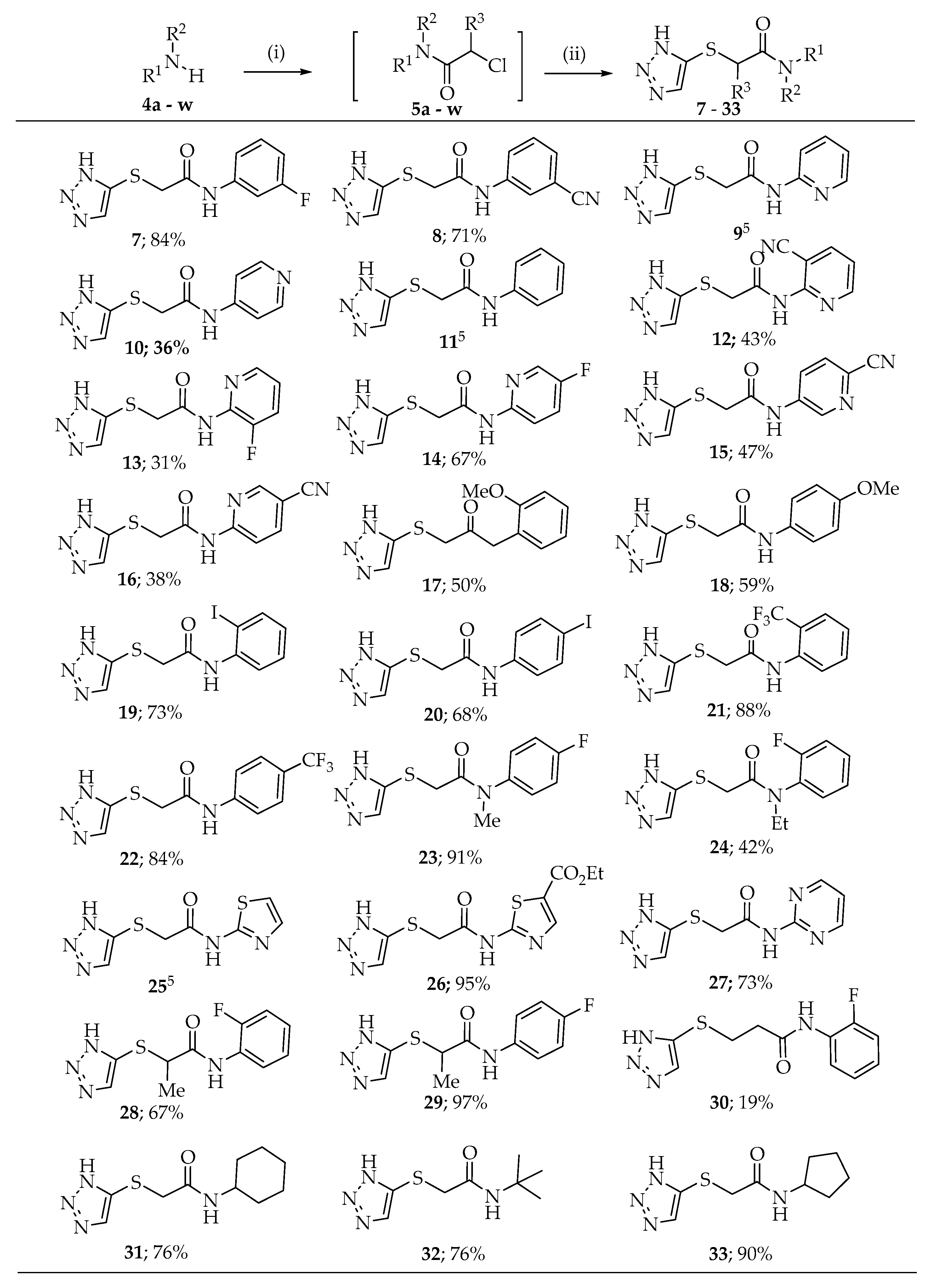
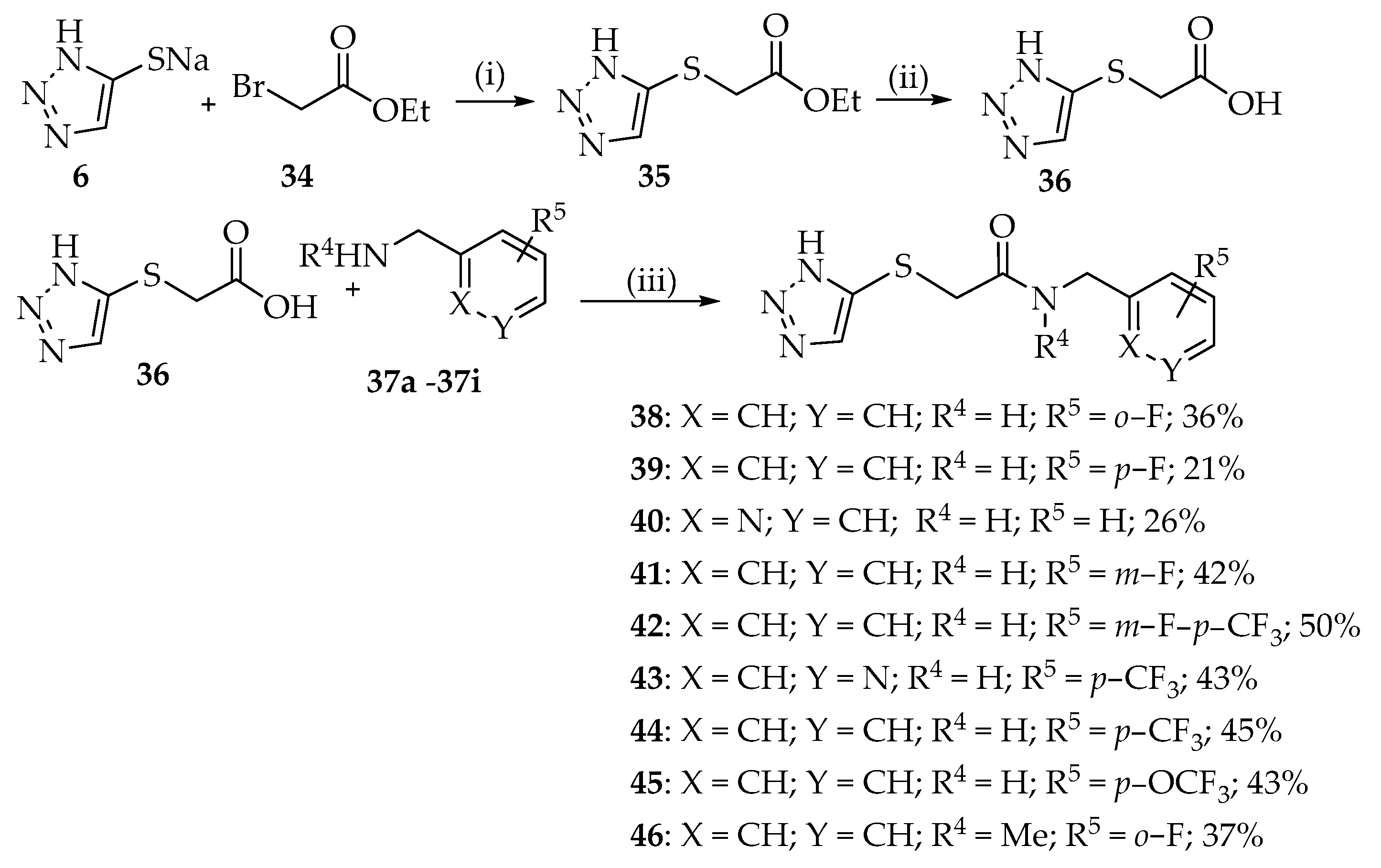
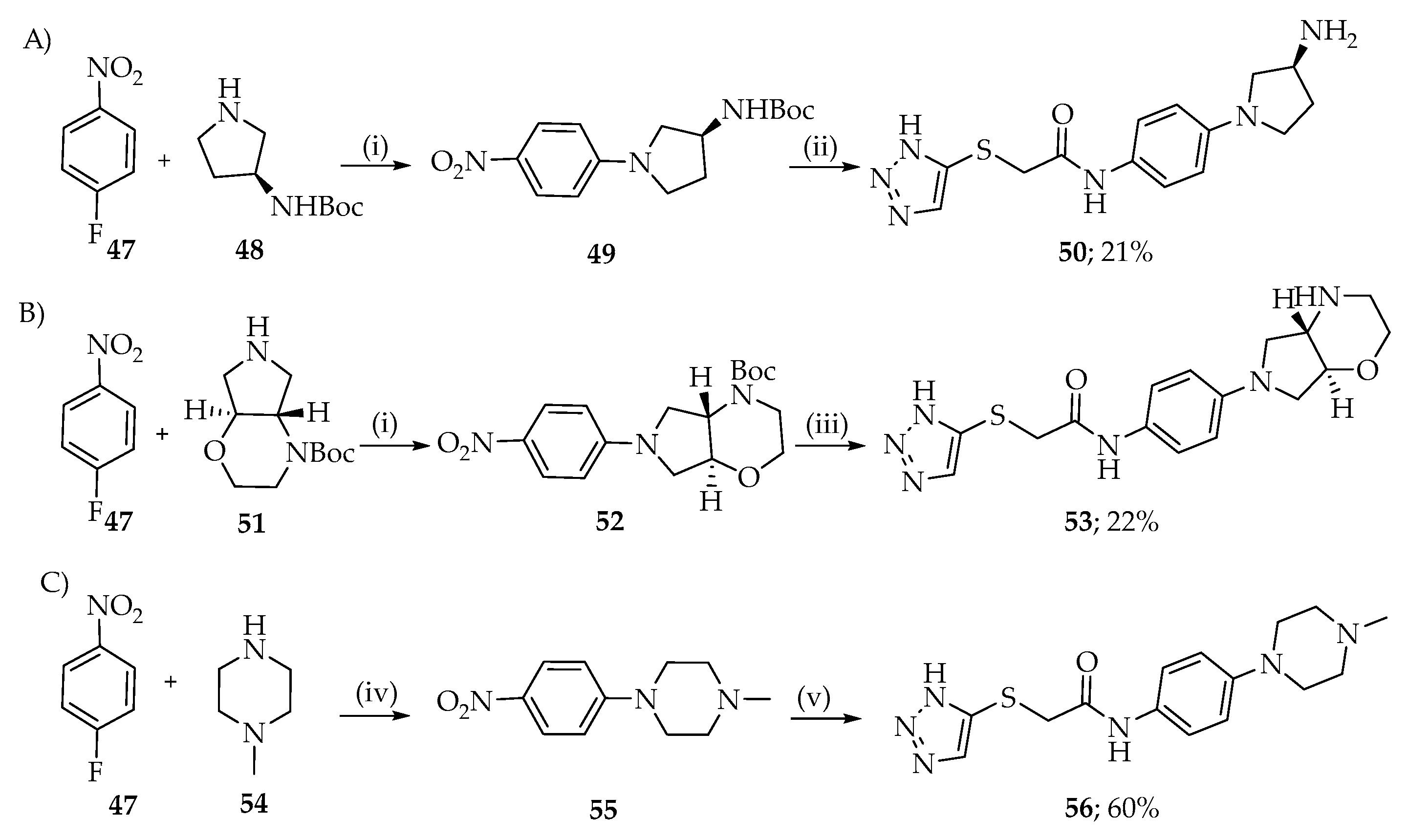
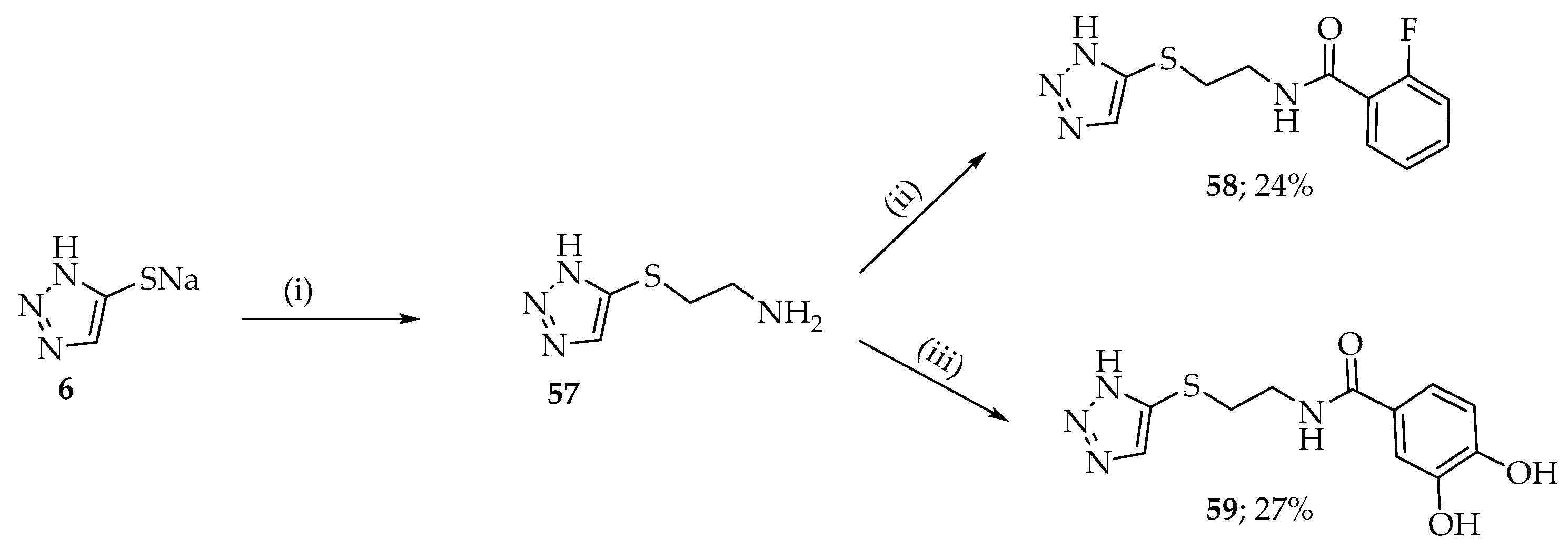
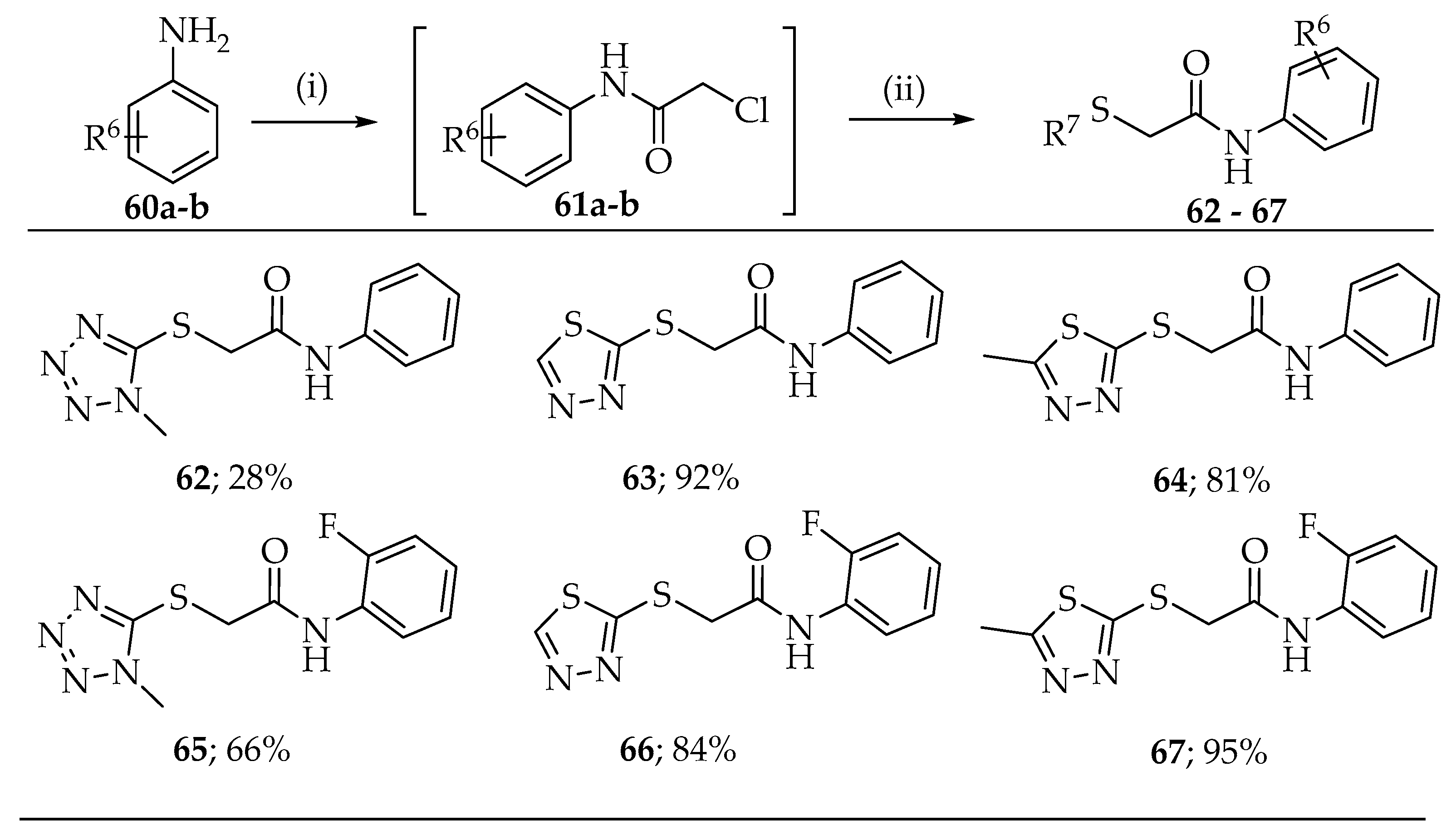
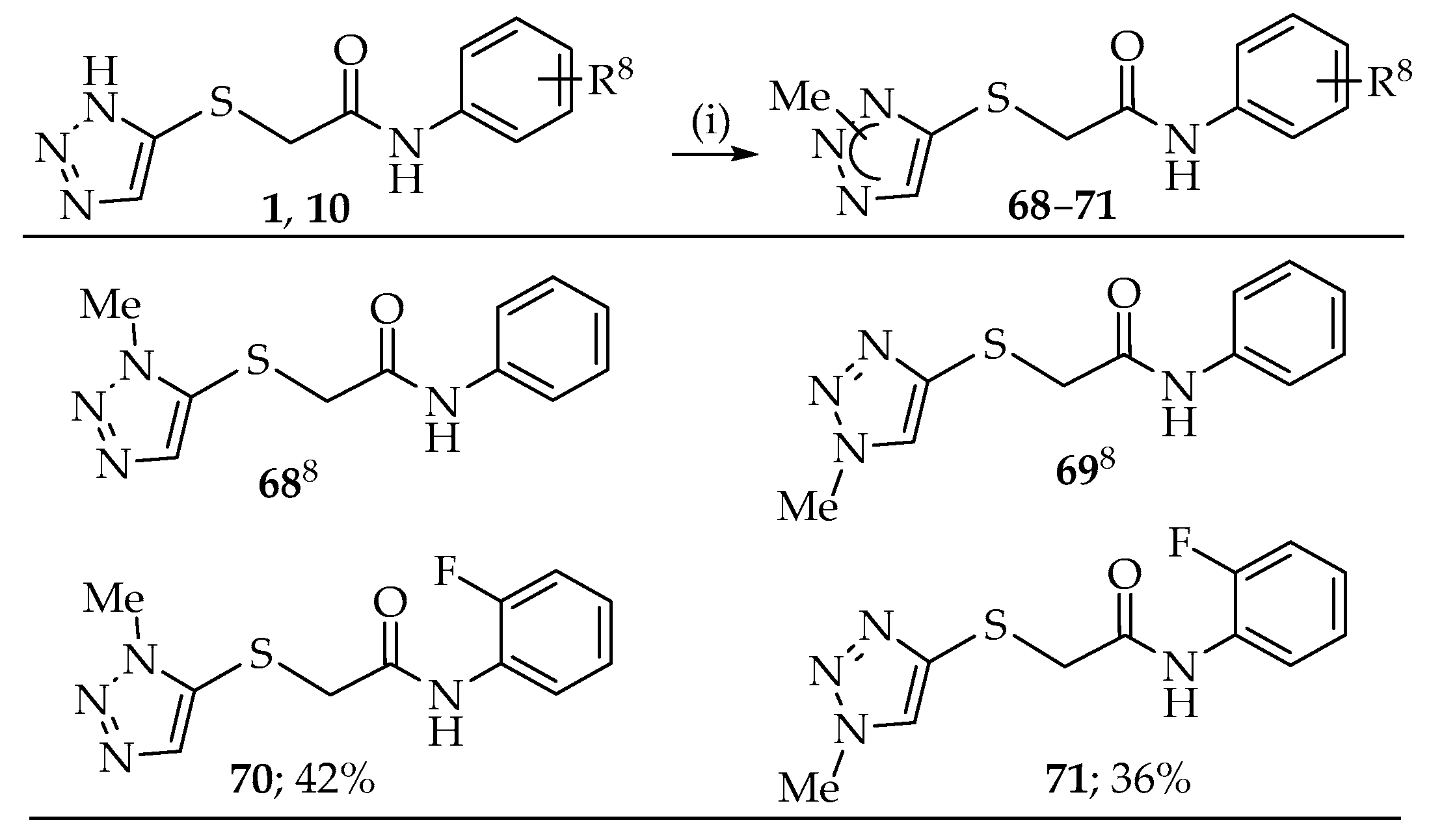
2.1. Synthesis
2.2. Antibacterial Studies
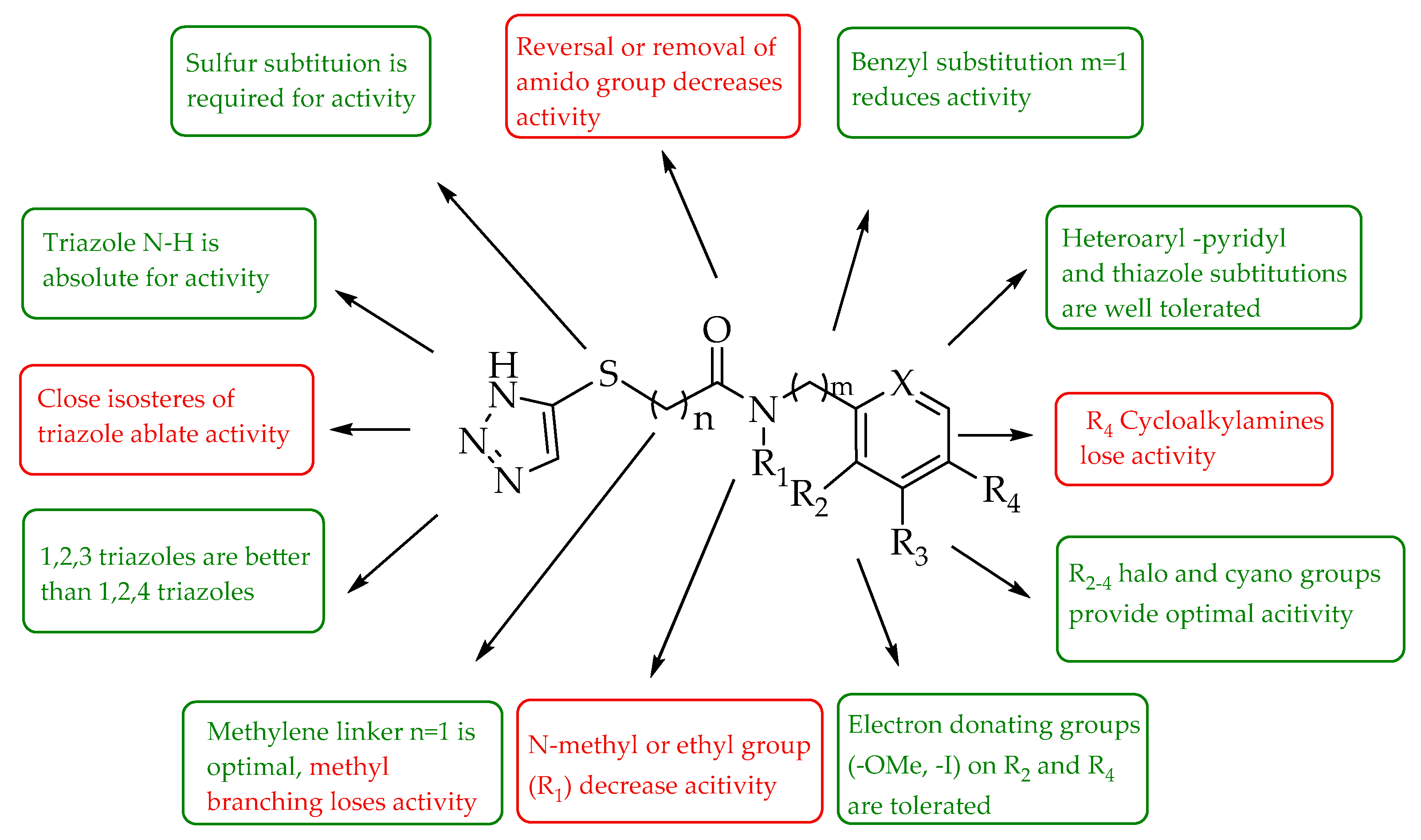
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Minimal Inhibitory Concentration Testing
4.3. Whole Cell Accumulation in E. coli K12
4.4. In Vitro ADME Profiling
4.5. Pharmacokinetic Evaluation in Mice
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spellberg, B.; Bartlett, J.; Wunderink, R.; Gilbert, D.N. Novel approaches are needed to develop tomorrow’s antibacterial therapies. Am. J. Respir. Crit. Care Med. 2015, 191, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Martens, E.; Demain, A.L. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017, 70, 520–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spera, A.M.; Esposito, S.; Pagliano, P. Emerging antibiotic resistance: Carbapenemase-producing enterobacteria. Bad new bugs, still no new drugs. Le Infez. Med. 2019, 27, 357–364. [Google Scholar]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.J.; Dharuman, S.; Fernando, D.M.; Reeve, S.M.; Gee, C.T.; Yao, J.; Griffith, E.C.; Phelps, G.A.; Wright, W.C.; Elmore, J.M.; et al. Discovery and Characterization of the Antimetabolite Action of Thioacetamide-Linked 1,2,3-Triazoles as Disruptors of Cysteine Biosynthesis in Gram-Negative Bacteria. ACS Infect. Dis. 2020, 6, 467–478. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. BioEssays News Rev. Mol. Cell. Dev. Biol. 2002, 24, 637–648. [Google Scholar] [CrossRef]
- Schnell, R.; Sriram, D.; Schneider, G. Pyridoxal-phosphate dependent mycobacterial cysteine synthases: Structure, mechanism and potential as drug targets. Biochim. Biophys. Acta 2015, 1854, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Balaji, B.S.; Dalal, N. An expedient and rapid green chemical synthesis of N-chloroacetanilides and amides using acid chlorides under metal-free neutral conditions. Green Chem. Lett. Rev. 2018, 11, 552–558. [Google Scholar] [CrossRef]
- Drysdale, M.J.; Pritchard, M.C.; Horwell, D.C. Rationally designed “dipeptoid” analogs of CCK. Acid mimics of the potent and selective non-peptide CCK-B receptor antagonist (CI-988). J. Med. Chem. 1992, 35, 2573–2581. [Google Scholar] [CrossRef]
- Hooper, D.C. Structure of grepafloxacin relative to activity and safety profile. Clin. Microbiol. Infect. 1998, 4, S15–S20. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.T.; Fernandes, P.B. Structure-activity relationships of the fluoroquinolones. Antimicrob. Agents Chemother. 1989, 33, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- VanderWel, S.N.; Harvey, P.J.; McNamara, D.J.; Repine, J.T.; Keller, P.R.; Quin, J.; Booth, R.J.; Elliott, W.L.; Dobrusin, E.M.; Fry, D.W.; et al. Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4. J. Med. Chem. 2005, 48, 2371–2387. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Wilkinson, S.R.; Szular, J.; Kaiser, M. Nitrotriazole-based acetamides and propanamides with broad spectrum antitrypanosomal activity. Eur. J. Med. Chem. 2016, 123, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Shadrick, W.R.; Wallace, M.J.; Wu, Y.; Griffith, E.C.; Qi, J.; Yun, M.K.; White, S.W.; Lee, R.E. Pterin-sulfa conjugates as dihydropteroate synthase inhibitors and antibacterial agents. Bioorg. Med. Chem. Lett. 2016, 26, 3950–3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlitni, S.; Ferruccio, L.F.; Brown, E.D. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat. Chem. Biol. 2013, 9, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.J.; Liu, R. Design and Syntheses of New Antibiotics Inspired by Nature′s Quest for Iron in an Oxidative Climate. Acc. Chem. Res. 2021, 54, 1646–1661. [Google Scholar] [CrossRef]
- Lang, Y.; Shah, N.R.; Tao, X.; Reeve, S.M.; Zhou, J.; Moya, B.; Sayed, A.R.M.; Dharuman, S.; Oyer, J.L.; Copik, A.J.; et al. Combating Multidrug-Resistant Bacteria by Integrating a Novel Target Site Penetration and Receptor Binding Assay Platform into Translational Modeling. Clin. Pharmacol. Ther. 2021, 109, 1000–1020. [Google Scholar] [CrossRef]
- Muñoz, K.A.; Hergenrother, P.J. Facilitating Compound Entry as a Means to Discover Antibiotics for Gram-Negative Bacteria. Acc. Chem. Res. 2021, 54, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Manchester, J.I.; Buurman, E.T.; Bisacchi, G.S.; McLaughlin, R.E. Molecular determinants of AcrB-mediated bacterial efflux implications for drug discovery. J. Med. Chem. 2012, 55, 2532–2537. [Google Scholar] [CrossRef]
- Zgurskaya, H.I. Introduction: Transporters, Porins, and Efflux Pumps. Chem. Rev. 2021, 121, 5095–5097. [Google Scholar] [CrossRef]
- Hatfield, M.J.; Umans, R.A.; Hyatt, J.L.; Edwards, C.C.; Wierdl, M.; Tsurkan, L.; Taylor, M.R.; Potter, P.M. Carboxylesterases: General detoxifying enzymes. Chem.-Biol. Interact. 2016, 259 Pt B, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, M.P. Performance Standards for Antimicrobial Susceptibility Testing, 30th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020; 332p. [Google Scholar]
- Parker, E.N.; Drown, B.S.; Geddes, E.J.; Lee, H.Y.; Ismail, N.; Lau, G.W.; Hergenrother, P.J. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol. 2020, 5, 67–75. [Google Scholar] [CrossRef]
- Geddes, E.J.; Li, Z.; Hergenrother, P.J. An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protoc. 2021, 16, 4833–4854. [Google Scholar] [CrossRef]
- Sharma, S.; Rao, R.; Reeve, S.M.; Phelps, G.A.; Bharatham, N.; Katagihallimath, N.; Ramachandran, V.; Raveendran, S.; Sarma, M.; Nath, A.; et al. Azaindole Based Potentiator of Antibiotics against Gram-Negative Bacteria. ACS Infect. Dis. 2021, 7, 3009–3024. [Google Scholar] [CrossRef]
- North, E.J.; Scherman, M.S.; Bruhn, D.F.; Scarborough, J.S.; Maddox, M.M.; Jones, V.; Grzegorzewicz, A.; Yang, L.; Hess, T.; Morisseau, C.; et al. Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg. Med. Chem. 2013, 21, 2587–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, E.C.; Zhao, Y.; Singh, A.P.; Conlon, B.P.; Tangallapally, R.; Shadrick, W.R.; Liu, J.; Wallace, M.J.; Yang, L.; Elmore, J.M.; et al. Ureadepsipeptides as ClpP Activators. ACS Infect. Dis. 2019, 5, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Y.; Wang, X.Y.; Wang, B.; He, H.Y.; Liu, J.Y.; Xiang, M.L.; He, J.; Wu, X.H.; Yang, L. Synthesis, preliminary structure-activity relationships, and in vitro biological evaluation of 6-aryl-3-amino-thieno[2,3-b]pyridine derivatives as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2013, 23, 2349–2352. [Google Scholar]
- Avdeef, A. High-Throughput Measurements of Solubility Profiles. In Pharmacokinetic Optimization in Drug Research; Wiley: Hoboken, NJ, USA, 2001; pp. 305–325. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | E. coli K12 MIC90 (μg/mL) a | Compound | Structure | E. coli K12 MIC90 (μg/mL) a | ||
|---|---|---|---|---|---|---|---|
| N.S. | +20 μg/mL Methionine | N.S. | +20 μg/mL Methionine | ||||
| 1 |  | 1.6 | 12.5 | 19 |  | 3.1 | n.d. |
| 2 |  | 3.1 | 12.5 | 20 |  | 6.3 | 25 |
| 3 |  | 3.1 | 12.5 | 21 |  | 25.0 | n.d. |
| 7 |  | 1.6 | 12.5 | 22 |  | 25 | n.d. |
| 8 |  | 3.1 | 12.5 | 23 |  | 25 | n.d. |
| 9 |  | 1.6 | 12.5 | 24 |  | 12.5 | 25 |
| 10 |  | 12.5 | 50 | 25 |  | 3.1 | 12.5 |
| 11 |  | 1.6 | 6.3 | 26 |  | 3.1 | 12.5 |
| 12 |  | 25.0 | n.d | 27 |  | 25 | n.d. |
| 13 |  | 6.3 | 25 | 28 |  | >200 | n.d. |
| 14 |  | 3.1 | 12.5 | 29 |  | 100 | n.d. |
| 15 |  | 6.3 | 50 | 30 |  | 6.3 | 25 |
| 16 |  | 3.1 | 6.3 | 31 |  | 25 | n.d. |
| 17 |  | 6.3 | n.d. | 32 |  | 12.5 | 25 |
| 18 |  | 3.1 | 12.5 | 33 |  | 12.5 | 25.0 |
| Item | Structure | E. coli K12 MIC90 (μg/mL) | |
|---|---|---|---|
| No Supplement | +20 μg/mL Methionine | ||
| 38 |  | 3.1 | n.d |
| 39 |  | 6.3 | 50 |
| 40 |  | 12.5 | >50 |
| 41 |  | 6.3 | n.d |
| 42 |  | 50 | n.d |
| 43 |  | 25 | n.d |
| 44 |  | 50 | n.d |
| 45 |  | 100 | n.d |
| 46 |  | 50 | n.d |
| Compound | Structure | E. coli K12 MIC90 (μg/mL) | Whole Cell Accumulation in E. coli (μM/1010 CFU) a |
|---|---|---|---|
| 50 |  | >200 | 9.0 ± 2 |
| 53 |  | >200 | 19 ± 1 |
| 56 |  | >200 | 4.7 ± 2 |
| 58 |  | 12.5 | 7.0 ± 1 |
| 59 |  | 50 | 1.0 ± 0.1 |
| Compound | Structure | E. coli K12 MIC90 (μg/mL) |
|---|---|---|
| 62 |  | >200 |
| 63 |  | >200 |
| 64 |  | >200 |
| 65 |  | >200 |
| 66 |  | >200 |
| 67 |  | >200 |
| 68 |  | >200 |
| 69 |  | >200 |
| 70 |  | >200 |
| 71 |  | >200 |
| Compounds | Avg. Sol a (µg/mL) | Plasma Stability (Mouse) t1/2 (h) | Plasma Stability (Human) t1/2 (h) | Metabolic Stability (Mouse) | Metabolic Stability (Human) | ||
|---|---|---|---|---|---|---|---|
| t1/2 (h) | Clint (mL/Min/Kg) | t1/2 (h) | Clint (mL/min/Kg) | ||||
| Verapamil | 39 ± 1.0 | 2.0 ± 0.1 | 0.4 ± 0.0 | 1.1 ± 0.1 | 51.1 | 1.5 ± 0.1 | 14.0 |
| 1 | 16 ± 0.8 | 0.39 ± 0.01 | 34.8 ± 0.3 | 0.27 ± 0.01 | 208.8 | 3.0 ± 0.2 | 7.0 |
| 2 | 18 ± 2.3 | 2.5 ± 0.2 | 36.3 ± 5.5 | 0.65 ± 0.03 | 87.4 | 4.7 ± 0.4 | 4.4 |
| 3 | 19 ± 1.5 | 1.10 ± 0.02 | 23.0 ± 1.1 | 1.13 ± 0.05 | 50.5 | 3.7 ± 0.3 | 5.7 |
| 9 | 20 ± 0.3 | 2.7 ± 0.2 | 19.9 ± 1.9 | 0.71 ± 0.02 | 80.4 | 2.7 ± 0.2 | 7.7 |
| 25 | 21 ± 1.8 | 13.3 ± 1.6 | 32.1 ± 5.4 | 2.0 ± 0.1 | 29.1 | 3.6 ± 0.3 | 5.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dharuman, S.; Wallace, M.J.; Reeve, S.M.; Bulitta, J.B.; Lee, R.E. Synthesis and Structure–Activity Relationship of Thioacetamide-Triazoles against Escherichia coli. Molecules 2022, 27, 1518. https://doi.org/10.3390/molecules27051518
Dharuman S, Wallace MJ, Reeve SM, Bulitta JB, Lee RE. Synthesis and Structure–Activity Relationship of Thioacetamide-Triazoles against Escherichia coli. Molecules. 2022; 27(5):1518. https://doi.org/10.3390/molecules27051518
Chicago/Turabian StyleDharuman, Suresh, Miranda J. Wallace, Stephanie M. Reeve, Jürgen B. Bulitta, and Richard E. Lee. 2022. "Synthesis and Structure–Activity Relationship of Thioacetamide-Triazoles against Escherichia coli" Molecules 27, no. 5: 1518. https://doi.org/10.3390/molecules27051518
APA StyleDharuman, S., Wallace, M. J., Reeve, S. M., Bulitta, J. B., & Lee, R. E. (2022). Synthesis and Structure–Activity Relationship of Thioacetamide-Triazoles against Escherichia coli. Molecules, 27(5), 1518. https://doi.org/10.3390/molecules27051518