
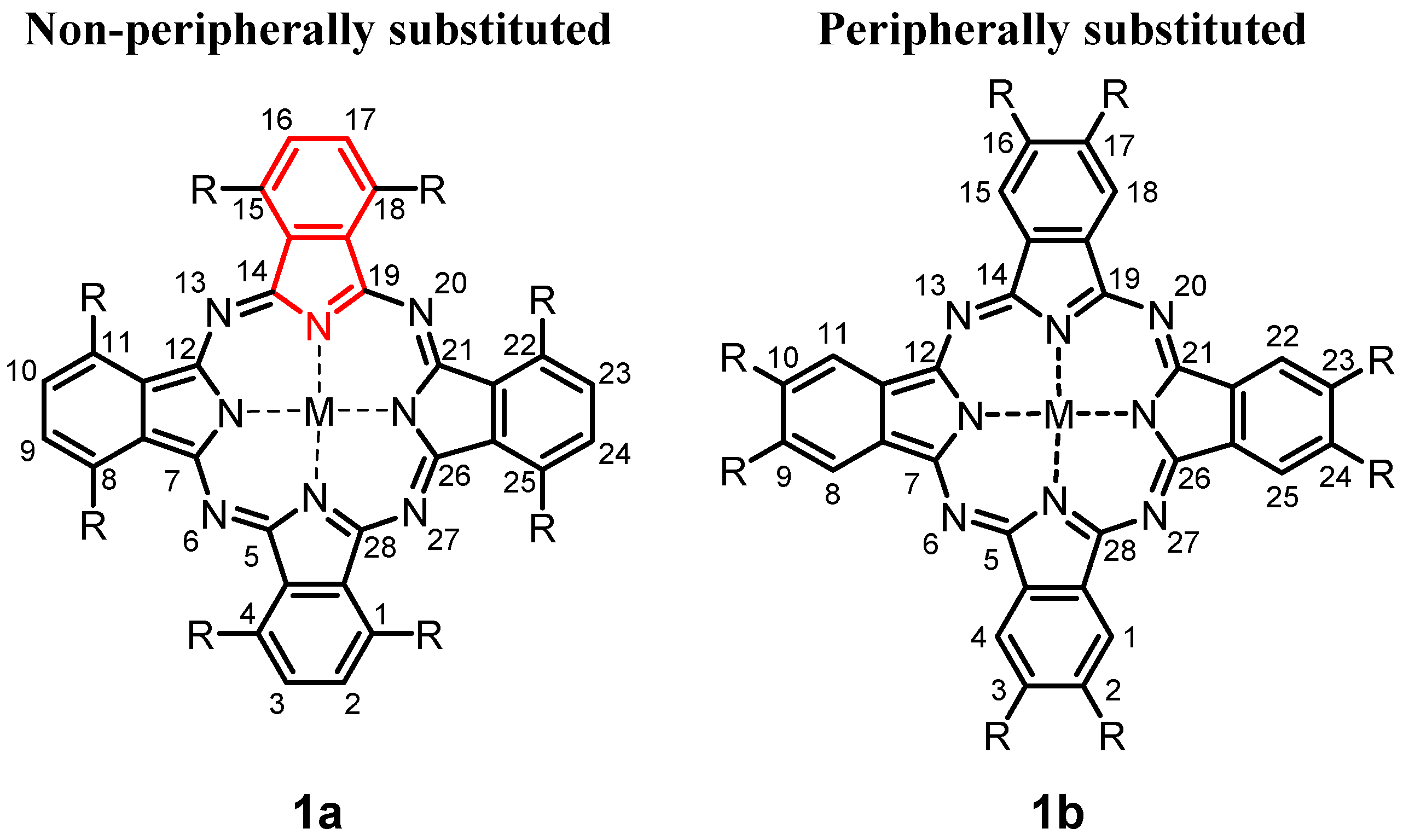
Octakis(dodecyl)phthalocyanines: Influence of Peripheral versus Non-Peripheral Substitution on Synthetic Routes, Spectroscopy and Electrochemical Behaviour


Abstract
:
1. Introduction
2. Results and Discussion
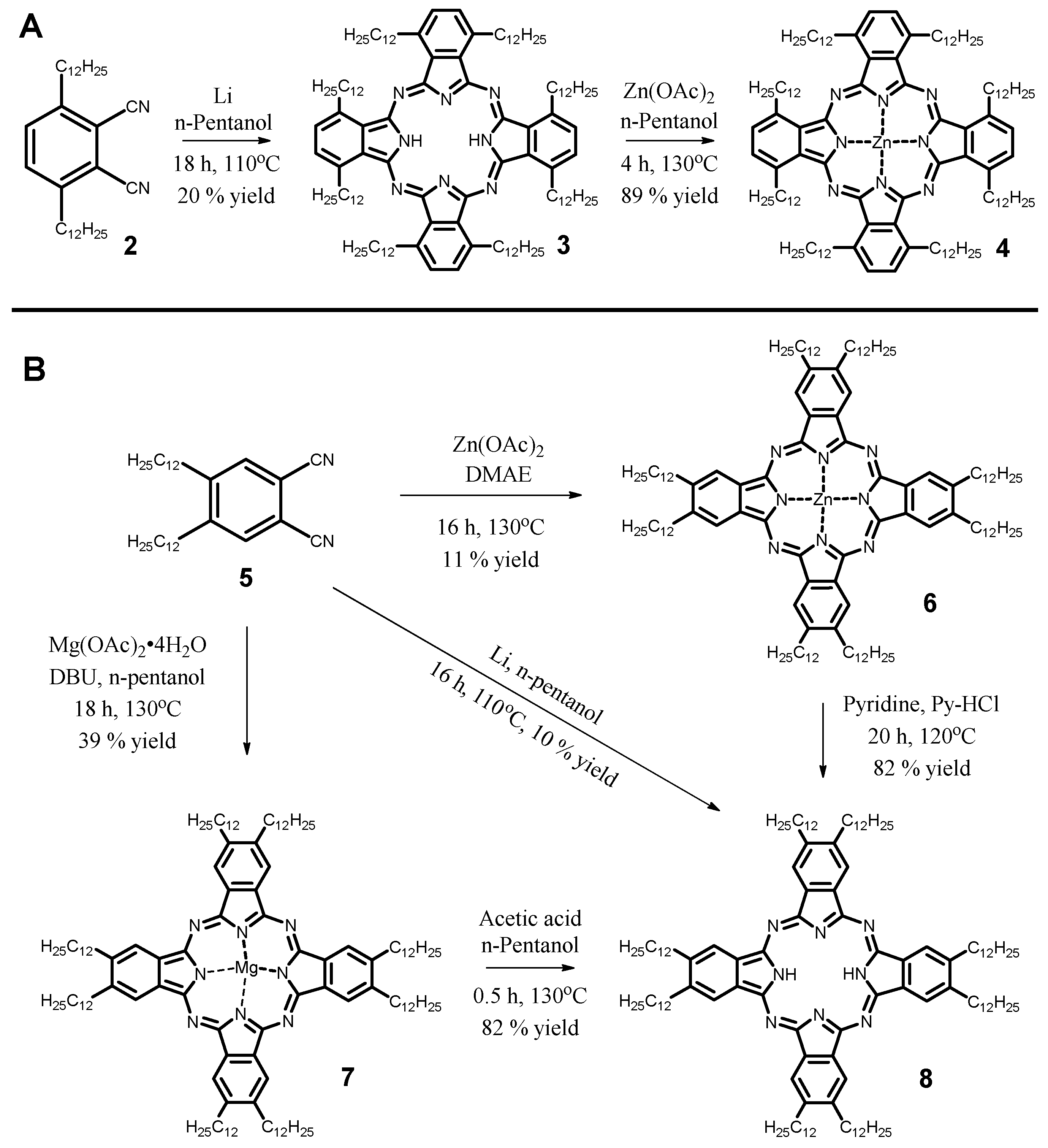
2.1. Synthesis
2.2. 1H NMR
2.3. FTIR–ATR and UV–Vis Spectroscopy
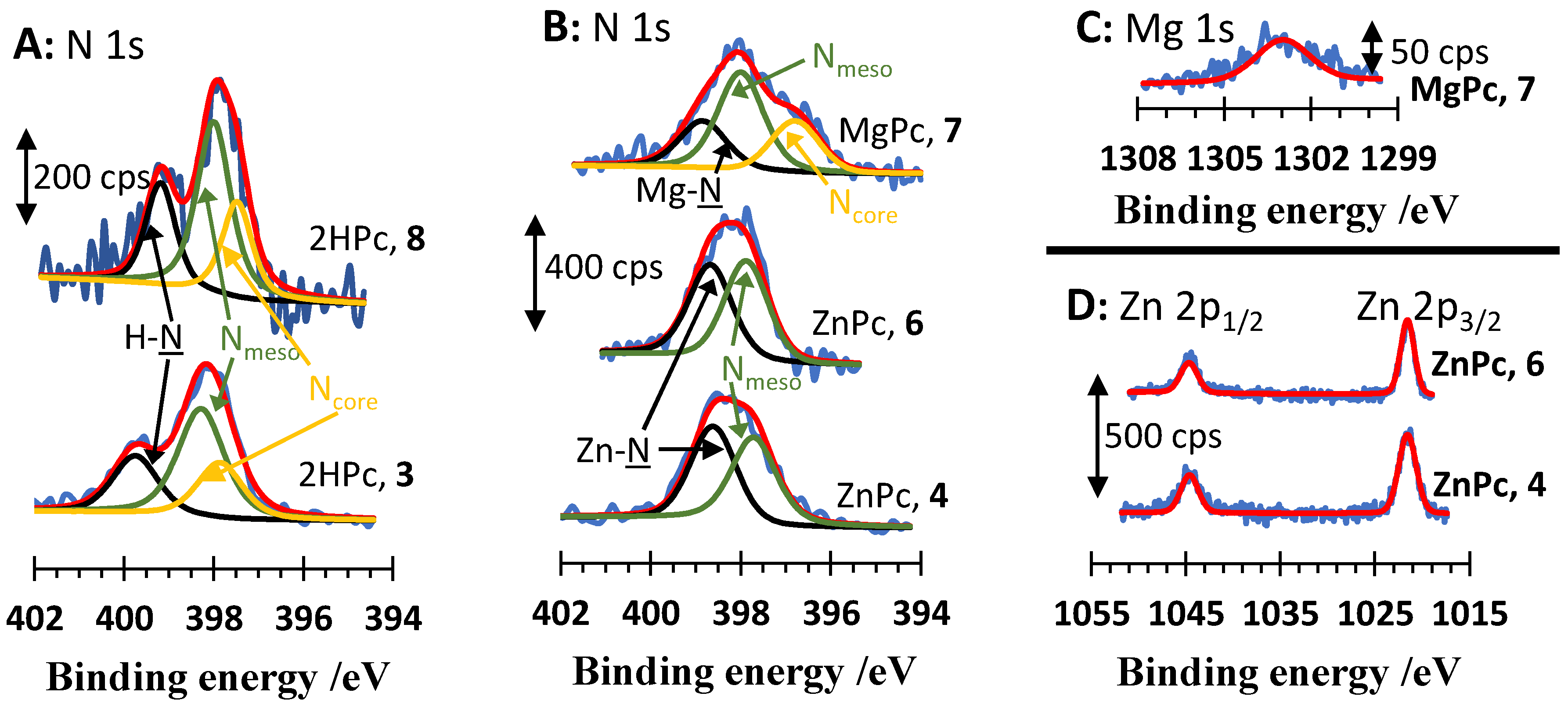
2.4. X-ray Photoelectron Spectroscopy
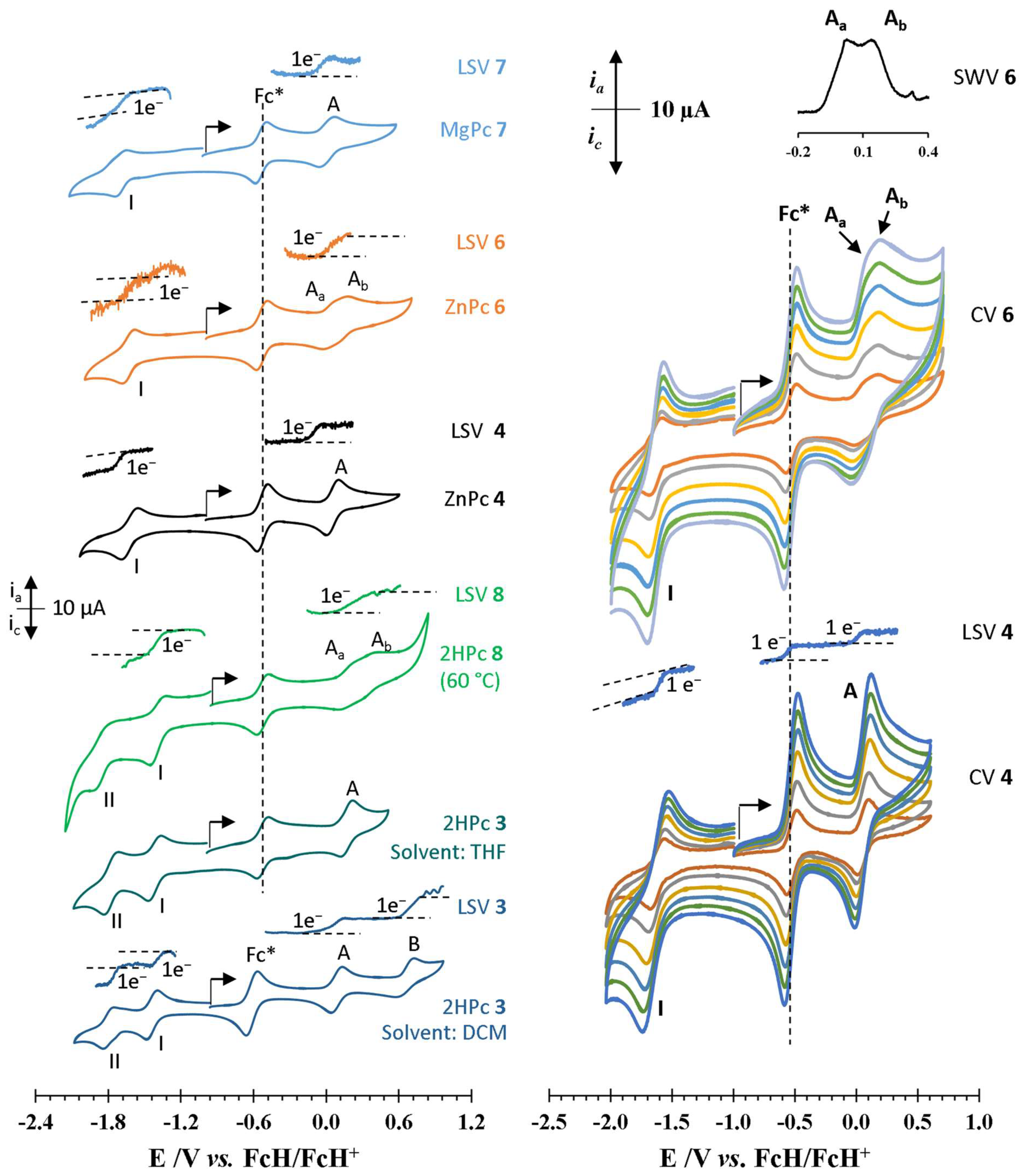
2.5. Electrochemistry
3. Materials and Methods
3.1. General Procedures
3.2. Spectroscopic Characterization Techniques
3.2.1. Nuclear Magnetic Resonance
3.2.2. UV–Vis Spectroscopy
3.2.3. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy
3.2.4. X-ray Photoelectron Spectroscopy
3.3. Synthesis
3.3.1. 1,4,8,11,15,18,22,25-Octakis(dodecyl)phthalocyanine, 3
3.3.2. 1,4,8,11,15,18,22,25-Octakis(dodecyl)phthalocyaninatozinc(II), 4
3.3.3. 2,3,9,10,16,17,23,24-Octakis(dodecyl)phthalocyaninatozinc(II), 6
3.3.4. 2,3,9,10,16,17,23,24-Octakis(dodecyl)phthalocyaninatomagnesium(II), 7
3.3.5. 2,3,9,10,16,17,23,24-Octakis(dodecyl)phthalocyanine, 8
3.4. Electrochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- De la Torre, G.; Nicolau, M.; Torres, T. Phthalocyanines: Synthesis, Supramolecular Organization, and Physical Properties. In Supramolecular Photosensitive and Electroactive Materials; Elsevier: Amsterdam, The Netherlands, 2001; pp. 1–111. ISBN 9780125139045. [Google Scholar]
- Nemykin, V.N.; Lukyanets, E.A. Synthesis of Substituted Phthalocyanines. Arkivoc 2010, 2010, 136–208. [Google Scholar] [CrossRef] [Green Version]
- Swarts, J.C.; Langner, E.H.G.; Krokeide-Hove, N.; Cook, M.J. Synthesis and Electrochemical Characterisation of Some Long Chain 1,4,8,11,15,18,22,25-Octa-Alkylated Metal-Free and Zinc Phthalocyanines Possessing Discotic Liquid Crystalline Properties. J. Mater. Chem. 2001, 11, 434–443. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Gągor, A.; Swarts, J.C. Electrochemical Evidence of Intramolecular Electronic Communication in Zr and Hf Phthalocyanines Bearing Ferrocene-Containing β-Diketonato Axial Ligands: Structure of [PcHf(FcCOCHCOC6H5)2]. Inorg. Chem. 2013, 52, 10245–10257. [Google Scholar] [CrossRef] [PubMed]
- Ince, M.; Martínez-Díaz, M.V.; Barberá, J.; Torres, T. Liquid Crystalline Phthalocyanine-Fullerene Dyads. J. Mater. Chem. 2011, 21, 1531–1536. [Google Scholar] [CrossRef]
- Fourie, E.; Swarts, J.C.; Chambrier, I.; Cook, M.J. Electrochemical and Spectroscopic Detection of Self-Association of Octa-Alkyl Phthalocyaninato Cadmium Compounds into Dimeric Species. Dalt. Trans. 2009, 7, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- L’Her, M.; Pondaven, A. Electrochemistry of Phthalocyanines. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Chapter 104; Academic Press: Cambridge, MA, USA, 2003; Volume 16, pp. 117–169. [Google Scholar]
- Van As, A.; Joubert, C.C.; Buitendach, B.E.; Erasmus, E.; Conradie, J.; Cammidge, A.N.; Chambrier, I.; Cook, M.J.; Swarts, J.C. Tetrabenzoporphyrin and -mono-, -cis-di- and Tetrabenzotriazaporphyrin Derivatives: Electrochemical and Spectroscopic Implications of Meso CH Group Replacement with Nitrogen. Inorg. Chem. 2015, 54, 5329–5341. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.J.; Chambrier, I.; White, G.F.; Fourie, E.; Swarts, J.C. Electrochemical and EPR Studies of Two Substituted Bis-Cadmium Tris-Phthalocyanine Complexes: Elucidation of Unexpectedly Different Free-Radical Character. Dalton Trans. 2009, 7, 1136–1144. [Google Scholar] [CrossRef]
- Apostol, P.; Bentaleb, A.; Rajaoarivelo, M.; Clérac, R.; Bock, H. Regiospecific Synthesis of Tetrasubstituted Phthalocyanines and Their Liquid Crystalline Order. Dalton Trans. 2015, 44, 5569–5576. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.M.S.; Kumari, P. Synthesis of Unsymmetrical Benzoporphyrazines in Functional Ionic Liquids and Formation of Self-Aggregates of Zinc(II) Pyridino[3,4]Tribenzoporphyrazines in Solutions. Tetrahedron 2009, 65, 2518–2524. [Google Scholar] [CrossRef]
- Cook, M.J.; Chambrier, I.; Cracknell, S.J.; Mayes, D.A.; Russell, D.A. Octa-Alkyl Zinc Phthalocyanines: Potential Photosensitizers for Use in the Photodynamic Therapy of Cancer. Photochem. Photobiol. 1995, 62, 542–545. [Google Scholar] [CrossRef]
- Venuti, E.; Della Valle, R.G.; Bilotti, I.; Brillante, A.; Cavallini, M.; Calò, A.; Geerts, Y.H. Absorption, Photoluminescence, and Polarized Raman Spectra of a Fourfold Alkoxy-Substituted Phthalocyanine Liquid Crystal. J. Phys. Chem. C 2011, 115, 12150–12157. [Google Scholar] [CrossRef]
- García-Iglesias, M.; Yum, J.H.; Humphry-Baker, R.; Zakeeruddin, S.M.; Péchy, P.; Vázquez, P.; Palomares, E.; Grätzel, M.; Nazeeruddin, M.K.; Torres, T. Effect of Anchoring Groups in Zinc Phthalocyanine on the Dye-Sensitized Solar Cell Performance and Stability. Chem. Sci. 2011, 2, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Ogata, H.; Nonaka, N.; Luk’yanets, E.A. Effect of Peripheral Substitution on the Electronic Absorption and Fluorescence Spectra of Metal-Free and Zinc Phthalocyanines. Chem. A Eur. J. 2003, 9, 5123–5134. [Google Scholar] [CrossRef] [PubMed]
- Detty, M.R.; Gibson, S.L.; Wagner, S.J. Current Clinical and Preclinical Photosensitizers for Use in Photodynamic Therapy. J. Med. Chem. 2004, 47, 3897–3915. [Google Scholar] [CrossRef] [PubMed]
- Torimtubun, A.A.A.; Follana-Berná, J.; Sánchez, J.G.; Pallarès, J.; Sastre-Santos, Á.; Marsal, L.F. Fluorinated Zinc and Copper Phthalocyanines as Efficient Third Components in Ternary Bulk Heterojunction Solar Cells. ACS Appl. Energy Mater. 2021, 4, 5201–5211. [Google Scholar] [CrossRef]
- Cao, R.; Thapa, R.; Kim, H.; Xu, X.; Kim, M.G.; Li, Q.; Park, N.; Liu, M.; Cho, J. Promotion of Oxygen Reduction by a Bio-Inspired Tethered Iron Phthalocyanine Carbon Nanotube-Based Catalyst. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biyiklioglu, Z.; Arslan, T.; Alawainati, F.A.; Manaa, H.; Jaffar, A.; Henari, F.Z. Comparative Nonlinear Optics and Optical Limiting Properties of Metallophthalocyanines. Inorg. Chim. Acta 2019, 486, 345–351. [Google Scholar] [CrossRef]
- Lee, W.; Yuk, S.B.; Choi, J.; Jung, D.H.; Choi, S.H.; Park, J.; Kim, J.P. Synthesis and Characterization of Solubility Enhanced Metal-Free Phthalocyanines for Liquid Crystal Display Black Matrix of Low Dielectric Constant. Dyes Pigments 2012, 92, 942–948. [Google Scholar] [CrossRef]
- Gericke, H.J.; Barnard, N.I.; Erasmus, E.; Swarts, J.C.; Cook, M.J.; Aquino, M.A.S. Solvent and Electrolyte Effects in Enhancing the Identification of Intramolecular Electronic Communication in a Multi Redox-Active Diruthenium Tetraferrocenoate Complex, a Triple-Sandwiched Dicadmium Phthalocyanine and a Ruthenocene-Containing β-Diketone. Inorg. Chim. Acta 2010, 363, 2222–2232. [Google Scholar] [CrossRef]
- Sharman, W.M.; Van Lier, J.E. Synthesis and Photodynamic Activity of Novel Asymmetrically Substituted Fluorinated Phthalocyanines. Bioconjug. Chem. 2005, 16, 1166–1175. [Google Scholar] [CrossRef]
- Terekhov, D.S.; Nolan, K.J.M.; McArthur, C.R.; Leznoff, C.C. Synthesis of 2,3,9,10,16,17,23,24-Octaalkynylphthalocyanines and the Effects of Concentration and Temperature on Their 1H NMR Spectra. J. Org. Chem. 1996, 61, 3034–3040. [Google Scholar] [CrossRef] [PubMed]
- Cammidge, A.N.; Chambrier, I.; Cook, M.J.; Hughes, D.L.; Rahman, M.; Sosa-Vargas, L. Phthalocyanine Analogues: Unexpectedly Facile Access to Non-Peripherally Substituted Octaalkyl Tetrabenzotriazaporphyrins, Tetrabenzodiazaporphyrins, Tetrabenzomonoazaporphyrins and Tetrabenzoporphyrins. Chem. A Eur. J. 2011, 17, 3136–3146. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.J. 1,4,8,11,15,18,22,25-Octasubstituted Phthalocyanines: The Contrasting Effects of Alkyl and Alkoxy Substituents on Molecular Self-Assembly. J. Mater. Sci. Mater. Electron. 1994, 5, 117–128. [Google Scholar] [CrossRef]
- Duro, J.A.; De la Torre, G.; Barbera, J.; Serrano, J.L.; Torres, T. Synthesis and liquid crystal behaviour of Metal-Free and Metal-Containing Phthalocyanines Substituted with Long-Chain Amide Groups. Chem. Mater. 1996, 8, 1061–1066. [Google Scholar] [CrossRef]
- Moreira, L.M.; Dos Santos, F.V.; Lyon, J.P.; Maftoum-Costa, M.; Pacheco-Soares, C.; Soares Da Silva, N. Photodynamic Therapy: Porphyrins and Phthalocyanines as Photosensitizers. Aust. J. Chem. 2008, 61, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Heslop, R.B.; Robinson, P.L. Inorganic Chemistry: A Guide to Advanced Study, 3rd ed.; Elsevier Publishing Company: Amsterdam, The Netherlands, 1967; p. 322. [Google Scholar]
- Swarts, P.J.; Swarts, J.C. Fundamentals and Applications in Solution-Phase Electrochemistry and Electrocatalysis. In Applications of Porphyrinoids as Functional Materials; Lang, H., Rueffer, T., Eds.; Royal Society of Chemistry: London, UK, 2021; Chapter 1; pp. 1–43. ISBN 978-1-83916-414-9. [Google Scholar]
- Yilmaz, I.; Burkut Koçak, M. Electrochemical, Spectroelectrochemical, and Pyridine Binding Properties of Tetrathia Macrocycle-Bridged Dimeric Cobalt Phthalocyanine. Polyhedron 2004, 23, 1279–1285. [Google Scholar] [CrossRef]
- Tolbin, A.Y.; Pushkarev, V.E.; Balashova, I.O.; Dzuban, A.V.; Tarakanov, P.A.; Trashin, S.A.; Tomilova, L.G.; Zefirov, N.S. A Highly Stable Double-Coordinated 2-Hydroxy-Tri(Tert-Butyl)-Substituted Zinc Phthalocyanine Dimer: Synthesis, Spectral Study, Thermal Stability and Electrochemical Properties. New J. Chem. 2014, 38, 5825–5831. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Erasmus, E.; Niemantsverdriet, J.W.; Swarts, J.C. Properties of Manganese(III) Ferrocenyl-β-Diketonato Complexes Revealed by Charge Transfer and Multiplet Splitting in the Mn 2p and Fe 2p X-Ray Photoelectron Envelopes. Molecules 2016, 21, 1427. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax/nm (logε) a | ||
|---|---|---|---|
| Q | Qx | Qy | |
| 2HPc, 3 | - | 694 (5.01) | 725 (5.08) |
| ZnPc, 4 | 698 (5.31) | - | - |
| ZnPc, 6 | 679 (5.29) | - | - |
| MgPc, 7 | 681 (5.50) | - | - |
| 2HPc, 8 | - | 668 (5.20) | 704 (5.25) |
| Compound | Nmeso 1s BE/eV (Atomic %) | N-M; Ncore BE/eV (Atomic %) | Metal 1s or 2p BE/eV (Atomic %) | Molecular Stoichiometry e Experimental (Theoretical) |
|---|---|---|---|---|
| 3 (M = 2H) | 398.3 (1.77) | 399.8 (0.88); 397.9 (0.88) | - | Nmeso:NH:Ncore = 4:2:2 f (4:2:2) g |
| 4 (M = Zn) | 397.7 (2.25) | 398.6 (2.25); - | 1044.6 a; 1021.6 b (0.55) c | Nmeso:NZn:Zn = 4:4:0.98 f (4:4:1) g |
| 6 (M = Zn) | 397.9 (2.39) | 398.7 (2.39); - | 1044.7 a; 1021.6 b (0.59) c | Nmeso:NZn:Zn = 4:4:0.99 f (4:4:1) g |
| 7 (M = Mg) | 398.0 (2.18) | 398.9 (1.09); 396.7 (1.09) | 1303.0 d (0.51) | Nmeso:NZn:Ncore:Mg = 4:2:2:0.94 f (4:2:2:1) g |
| 8 (M = 2H) | 398.0 (1.23) | 399.2 (0.61); 397.5 (0.61) | - | Nmeso:NH:Ncore = 4:2:2 f (4:2:2) g |
| Wave | Epa/V | ΔEp/mV a | E°′/V | ipa/μA | ipc/ipa | Wave | Epa/V | ΔEp/mV a | E°′/V | ipa/μA | ipc/ipa |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Decamethylferrocene (THF) | Ferrocene (THF) | ||||||||||
| - | −0.488 | 76 | −0.527 | 6.00 | 0.96 | - | 0.039 | 78 | 0.000 | 10.80 | 0.98 |
| Decamethylferrocene (DCM) | Ferrocene (DCM) | ||||||||||
| - | −0.575 | 74 | −0.614 | 5.00 | 0.98 | - | 0.035 | 70 | 0.000 | 6.50 | 0.96 |
| 3 (2H, non-peripheral, DCM) ΔE°′HL = 1.531 V b | 8 (2H, peripheral, THF) ΔE°′HL = 1.562 V b | ||||||||||
| A | 0.136 | 97 | 0.088 | 4.75 | 1.00 | Aa | 0.227 f | 111 | 0.172 | 1.56 | 0.83 |
| B | 0.726 | 143 | 0.655 c | 3.38 | 0.56 | Ab | 0.416 f | 98 | 0.367 | 1.80 | 0.83 |
| I | −1.396 | 93 | −1.443 | 4.25 d | 0.97 e | I | −1.326 | 127 | −1.390 | 3.44 d | 0.82 e |
| II | −1.754 | 92 | −1.800 | 3.13 d | 0.96 e | II | −1.785 | 135 | −1.853 | 2.50 d | 1.00 e |
| 3 (2H, non-peripheral, THF) ΔE°′HL = 1.590 V b | 6 (Zn, peripheral, THF) ΔE°′HL = 1.657 V b | ||||||||||
| A | 0.221 | 99 | 0.172 | 3.40 | 0.90 | Aa | 0.067 f | 86 | 0.024 | 1.90 | 0.93 |
| I | −1.364 | 107 | −1.418 | 3.40 d | 0.88 e | Ab | 0.181 f | 78 | 0.142 | 1.67 | 1.00 |
| II | −1.716 | 121 | −1.777 | 3.40 d | 0.88 e | I | −1.578 | 113 | −1.633 | 3.10 d | 0.85 e |
| 4 (Zn, non-peripheral, THF) ΔE°′HL = 1.673 V b | 7 (Mg, peripheral, THF) ΔE°′HL = 1.705 V b | ||||||||||
| A | 0.105 | 105 | 0.053 | 3.60 | 0.97 | A | 0.080 | 135 | 0.012 | 2.40 | 0.96 |
| I | −1.556 | 128 | −1.620 | 2.60 d | 1.00 e | I | −1.646 | 95 | −1.693 | 1.90 d | 0.95 e |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swart, G.; Fourie, E.; Swarts, J.C. Octakis(dodecyl)phthalocyanines: Influence of Peripheral versus Non-Peripheral Substitution on Synthetic Routes, Spectroscopy and Electrochemical Behaviour. Molecules 2022, 27, 1529. https://doi.org/10.3390/molecules27051529
Swart G, Fourie E, Swarts JC. Octakis(dodecyl)phthalocyanines: Influence of Peripheral versus Non-Peripheral Substitution on Synthetic Routes, Spectroscopy and Electrochemical Behaviour. Molecules. 2022; 27(5):1529. https://doi.org/10.3390/molecules27051529
Chicago/Turabian StyleSwart, Glendin, Eleanor Fourie, and Jannie C. Swarts. 2022. "Octakis(dodecyl)phthalocyanines: Influence of Peripheral versus Non-Peripheral Substitution on Synthetic Routes, Spectroscopy and Electrochemical Behaviour" Molecules 27, no. 5: 1529. https://doi.org/10.3390/molecules27051529
APA StyleSwart, G., Fourie, E., & Swarts, J. C. (2022). Octakis(dodecyl)phthalocyanines: Influence of Peripheral versus Non-Peripheral Substitution on Synthetic Routes, Spectroscopy and Electrochemical Behaviour. Molecules, 27(5), 1529. https://doi.org/10.3390/molecules27051529