Abstract
Several sterically protected, low-coordinate organophosphorus compounds with P=P, P=C, and C≡P bond are described in this study. Molecules such as diphosphenes, phosphaalkenes, 1-phosphaallenes, 1,3-diphosphaallenes, 3,4-diphosphinidenecyclobutenes, and phosphaalkynes are stabilized with an extremely bulky 2,4,6-tri-t-butylphenyl (Mes*) group. The synthesis, structures, physical, and chemical properties of these molecules are discussed, together with some successful applications in catalytic organic reactions.
1. Introduction
Low-coordinate organophosphorus compounds with coordination numbers of 1 or 2 were once believed to only exist as unstable species. However, by introducing a sterically bulky group such as 2,4,6-tri-t-butylphenyl into a molecule, various kinds of unusual phosphorus compounds have been isolated as kinetically stable species since 1981. In this review, the characteristics of several compounds carrying P=P, P=C, and P≡C bonds—including the preparation, structural, physical, theoretical and chemical aspects, as well as some catalytic applications to organic reactions—are described.
2. Steric Protection and the First “Phosphobenzene”
2.1. Steric Protection for Stabilization of Unstable Compounds
As shown in Scheme 1, Okazaki and Inamoto [1] found that 2,4,6-tri-t-butylaniline (1) is oxidized with 2 moles of perbenzoic acid in dichloromethane to yield the corresponding nitrosobenzene (2), which is stable in air at room temperature and can be purified by column chromatography with alumina. In contrast to normal nitroso compounds such as unsubstituted nitrosobenzene, compound 2 is emerald green and does not dimerize to 1,3,2,4-dioxadiazetidine (3) either in solution or the solid state due to the steric hindrance. In general, the steric protection technique seems promising for the stabilization of unstable compounds.
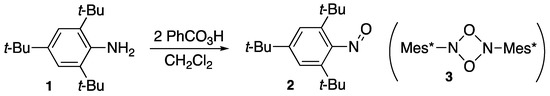
Scheme 1.
A sterically protected nitrosobenzene (2); Mes* = 2,4,6-t-Bu3C6H2.
In an attempt to introduce a phosphorus moiety to the phenyl ring, phosphorus trichloride was allowed to react with 1,3,5-tri-t-butylbezene (4) in the presence of aluminum chloride. As reported by Cook [2], in place of the expected 2,4,6-tri-t-butylphenylphosphinic chloride (5), after partial hydrolysis, t-butyl-3,5-di-t-butylphenylphosphinic chloride (6) was obtained, indicating that one of the t-butyl groups migrated from the aromatic ring to the phosphorus atom under Friedel–Crafts reaction conditions, as shown in Scheme 2 [3]. It turned out that t-butyl is prone to move around on the ring to rearrange the original positions under the strong acidic conditions, probably due to a labile t-butyl cation, e.g., 7 and 8. Therefore, another strategy was needed to introduce a phosphorus functional group on a bulky benzene nucleus to utilize 2,4,6-tri-t-butylphenyl as a sterically demanding group (hereafter abbreviated as Mes*).
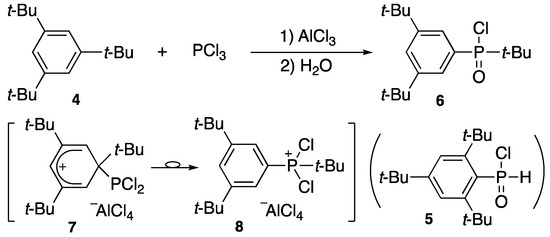
Scheme 2.
Friedel–Crafts-type reaction of 2,4,6-tri-t-butylbenzene (4).
2.2. Preparation and Characterization of Diphosphenes
“Phosphobenzene (9)” was reported by Köhler and Michaelis [4] as the phosphorus analogue of azobenzene. Later, however, it was revealed that the structure of this compound 9 comprised oligomers such as 10 [5,6], 11 [7,8], and 12 [9,10] according to the molecular weight determination and X-ray analysis (Scheme 3). Since then, it has been believed that such compounds with double bonds involving heavier main group elements never existed as stable compounds.
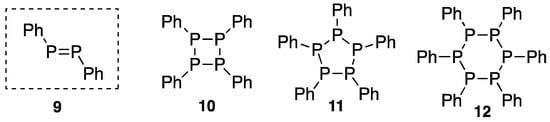
Scheme 3.
“Phosphobenzene”.
As shown in Scheme 4, 1,3,5-tri-t-butylbezene (4) is brominated to yield 1-bromo-2,4,6-tri-t-butylbenzene (13) and is converted to 2,4,6-tri-t-butylphenylphosphonous dichloride (15) after a halogen–metal exchange with butyllithium, thus yielding 14 followed by the addition of phosphorus trichloride. Dichloride 15 reacts with lithium aluminum hydride to yield 2,4,6-tri-t-butylphenylphosphane (16). It should be noted that both bulky phosphorus compounds 15 and 16 are stable, easy to handle in air at room temperature, and can serve as appropriate starting materials for the study of low-coordinate organophosphorus compounds [11].
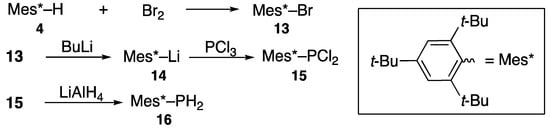
Scheme 4.
Preparation of sterically bulky phosphonous dichloride 15 and phosphane 16.
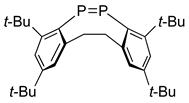
As shown in Scheme 5, dichloride 15 is allowed to react with magnesium in THF to yield a stable diphosphene 17 as an orange red crystalline material with an mp of 175–176 °C [12]. Some selected physical and X-ray data are listed in Table 1. It is noteworthy that (E)-bis(2,4,6-tri-t-butylphenyl)diphosphene (17), which turned out to be a true “phosphobeneze,” has a short P–P bond distance (2.034(2) Å) and shows a low 31P NMR chemical shift (δP 492.4 ppm).
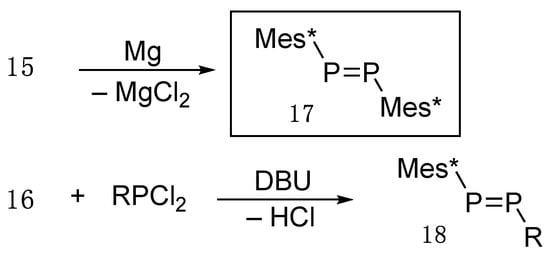
Scheme 5.
Preparation of diphosphenes (17 and 18); DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.

Table 1.
Some physical data of diphosphene 17.
According to the crystallographic analysis, the bond distance between the two phosphorus atoms is about 10% shorter than the regular P–P single bond, and the dihedral angle ∠C–P–P–C reveals a planar molecular system with (E)-configuration.
However, in our initial paper [12], we misreported the 31P NMR chemical shift of 17 as δP −59.00 ppm and immediately corrected the value for 17 to 492.4 ppm (C6D6) because it is one of the most important physical characteristics [13]. In the meantime, Lappert [15] critcized our initially reported chemical shift but Cowley [16] argued our structure of diphosphene itself as diphosphane Mes*P(H)–P(H)Mes* (31P NMR δP of −64.4 ppm for dl and δP of −65.0 ppm for meso) [17], despite our unambiguously determined X-ray analysis [12]. On the other hand, it turned out that the coupling constant between two phosphorus nuclei, 1JPP, is large and has been confirmed by NMR measurement for unsymmetrical diphosphene 18 prepared from the dehydrochlorination reaction of the primary phosphane 16 and RPCl2 with a base such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) [18] (Scheme 5, second row). Some selected coupling constants and chemical shifts are listed in Table 2 [19].
Table 2.
31P NMR data for some diphosphenes.
31P NMR data for (Z)-diphosphenes 21 (δP 368 ppm) and 22 (δP 394 ppm) show a higher chemical shift than (E)-diphosphenes. As shown in Scheme 6, the formation of (Z)-bis(2,4,6-tri-t-butylphenyl)diphosphene (21) is observed during the temperature- and wavelength-depending photolysis of (E)-diphosphene 17. The irradiation of 17 with a Hg lamp through a Pyrex filter at –40 °C [26], or argon-laser irradiation (514.5 nm) at −78 °C [27] yields an E/Z equilibrium mixture that returns to E-form upon warming, showing that (Z)-diphosphene 21 is thermally unstable due to the steric congestion caused by the two adjacent Mes* groups. Caminade reported the first order rate constant of E/Z isomerization reaction as ΔG≠273 = 20.35 kcal/mol based on 31P-NMR studies. When the photolysis is conducted without a Pyrex filter, regardless of the temperature between −78 °C and 0 °C [26], intramolecular insertion to a neighboring methyl group occurs and yields benzophosphaindane 24, most likely suggesting the generation of phosphinidene intermediate 23 during the photolysis.
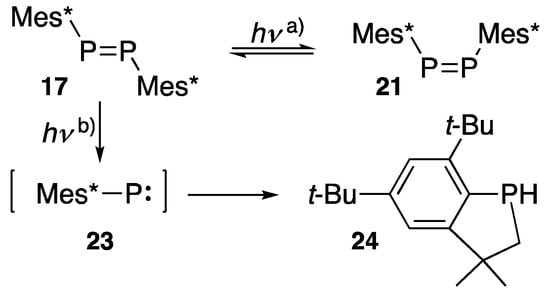
Scheme 6.
Temperature- and wavelength-dependent photolysis of diphosphene 17: (a) Hg lamp irradiation through a Pyrex filter or Ar-laser irradiation at low temperature; (b) Hg lamp without a Pyrex filter.
In the course of the synthetic route from 15 to 17 (Scheme 5, first row), a “free” 23 does not seem to be generated since Protasiewicz failed to obtain 17 under controlled reaction conditions such as when employing extremely dried THF with activated magnesium metal [29], instead yielding 24.
Scheme 7 shows that (Z)-diphosphene 22 is prepared via photolytic ring closing metathesis (RCM) on bis(E-diphosphene) 25 [28] with a Xe-lamp in benzene for 30 min at room temperature, though the yield of 22 is not satisfactory.
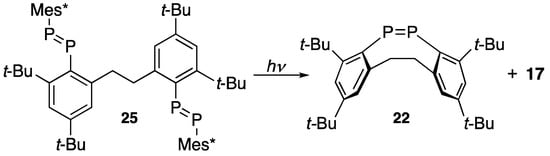
Scheme 7.
Ring closing metathesis of bis(diphosphene) 25 by Xe-lamp irradiation.
It should be noted that in the year of 1981, when a stable diphosphene 17 was isolated for the first time, West reported the successful isolation of a stable disilene (Mes2Si = SiMes2; Mes = 2,4,6-Me3C6H2) protected with four mesityl groups [30]. The molecular structures of these two compounds are simple but unusual and uncommon based on the traditional knowledge of organic, inorganic, and/or physical chemistry [31,32,33,34,35,36,37]. Since then, however, various kinds of sterically protected stable low-coordinate phosphorus compounds have been successfully prepared and characterized. Many sophisticated or simple protective groups other than Mes* with stronger or weaker steric ability, with or without electronic effect, or for special or common purposes have been developed, and some early examples have already been reviewed [18,38,39].
2.3. Chemical Reactivity of Diphosphenes
Diphosphenes demonstrate a variety of chemical reactions including photolysis, oxidation, reduction, hydrolysis, sulfurization, carbene-addition, and coordination to transition-metals [31,32]. In this section, selected examples are described in more detail.
Diphosphene 17 is allowed to react with sulfur in triethylamine at room temperature overnight to yield a stable diphosphene mono-sulfide 26, and the desulfurization to the starting diphosphene 17 is carried out with hexamethylphosphorous triamide in benzene at room temperature for 2 h. Sulfide 26 is converted to thiadiphosphirane 27 upon the irradiation of light with a medium-pressure Hg lamp at 0 °C for 5 min or heating in toluene for 30 min at 95 °C (Scheme 8) [40,41].
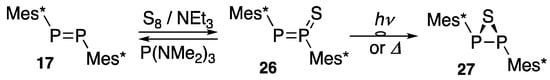
Scheme 8.
Reaction of diphosphene 17 with sulfur.
Unsymmetrical diphosphene 18B is allowed to react with (THF)Cr(CO)5 to yield an end-on complex 28 as a result of coordination at the less hindered site. However, complex 28 is converted to isomer 29 upon the irradiation of light with a medium-pressure Hg lamp in hexane at 0 °C for 5 min due to the E/Z isomerization around the P=P bond. The Z-ligand on chromium in 29 is thermally stable, probably because the steric environment reduces the enthalpy change between 28 and 29, in contrast to that of free ligand 18B (Scheme 9) [42].
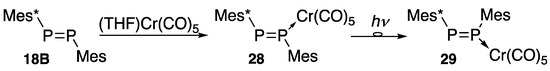
Scheme 9.
Reaction of diphosphene 18B with carbonylchromium(0); Mes = 2,4,6-Me3C6H2.
3. Stable Phosphaalkenes and Phosphaalkynes
3.1. Stable Phosphaalkenes and the Related Compounds
In 1966, Märkl reported the interesting preparation of a stable phosphorus-containing heterocyclic compound from 2,4,6-triphenylpyrylium tetrafluoroborate (30) [43]. The obtained triphenylphosphabenzene (or phosphinine) (31) contains a P=C bond in its canonical form, but the stability is estimated to be due to delocalization or aromaticity (Scheme 10).
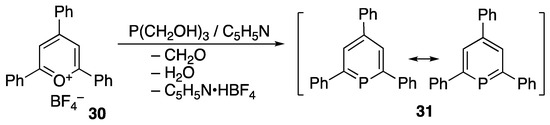
Scheme 10.
Preparation of phosphabenzene 31.
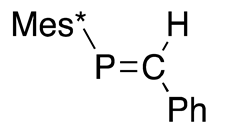
Later in 1978, Bickelhaupt reported a thermally stable phosphaalkene 32 with a localized P=C double bond that is sterically protected with mesityl group [44], as shown in Scheme 11. This successful isolation of 32 triggered our interest in the field of sterically protected, low-coordinate phosphorus compounds.
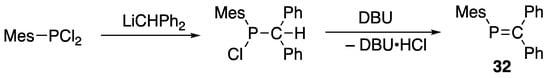
Scheme 11.
Preparation of phosphaethene 32; Mes = 2,4,6-Me3C6H2; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
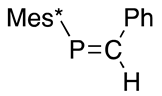
Alternatively, more sterically protected phosphaethenes with Mes* around phosphorus can be prepared by another method such as the phospha-Peterson reaction starting from silylphosphide and carbonyl compounds, thus successively preparing (E)-2-phenyl-1-(2,4,6-tri-t-butylphenyl)-1-phosphaethene (33), as shown in Scheme 12 [45].
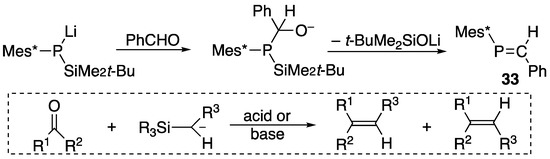
Scheme 12.
Preparation of phosphaalkene 33 and the Peterson reaction.
Interestingly, phosphaethene 33 in the (E)-form is isomerized in benzene upon the irradiation of light with a 100 W medium pressure Hg lamp at 0 °C for 6 h to yield an equilibrium mixture (3:7) with the corresponding (Z)-phosphaethene 34 [45], as shown in Scheme 13. After separation with silica-gel column chromatography, both isomers were analyzed by X-ray crystallography [46], and it was found that separated (Z)-phosphaethene 34 is thermally stable and does not isomerize to 33 even it is heated at 100 °C in toluene for 24 h.
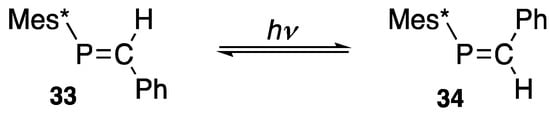
Scheme 13.
Photoisomerization of a phosphaethene.
Table 3 shows 31P-NMR chemical shifts for some selected phosphaalkenes. Aromatic phosphorus in 31 shows a higher shift than isolated phosphaalkenes, and (Z)-isomer 34 shows a slightly higher chemical shift than (E)-isomer 33.
Table 3.
31P NMR for some phosphaalkenes, phosphaalkynes, and related compounds.
Alternatively, Appel reported a convenient method for the preparation of useful 2-halo-1-(2,4,6-tri-t-butylphenyl)phosphaethene (35) starting from 2,4,6-tri-t-butylphenylphosphane (16) and trihalomethanes, as shown in Scheme 14 [57].
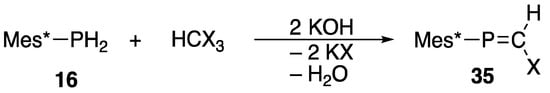
Scheme 14.
Preparation of 2-halophosphaethene 35.
3.2. Some Reactions of Phosphaalkenes
The Peterson method can be applied to prepare 3-phenyl-1-(2,4,6-tri-t-butylphenyl)-1-phosphaallene (36), as shown in Scheme 15 [47], indicating that the extended P=C system (phosphacumulenes) is also stable once sterically protected.
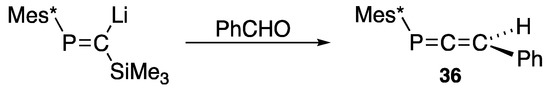
Scheme 15.
Preparation of phosphaallene 36 via the Peterson reaction.
On the other hand, when a silylphosphide is allowed to react with half of an equivalent of carbon dioxide, 1,3-bis(2,4,6-tri-t-butylphenyl)-1,3-diphosphaallene (37) is formed [48,58] via the phospha-Peterson reaction, as shown in Scheme 16 [47].
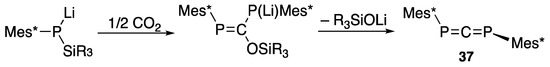
Scheme 16.
Preparation of diphosphaallene 37 with the phospha-Peterson reaction.
It is interesting to note that diphosphaallene 37 can be alternatively prepared from diphosphene 17 and dichlorocarbene followed by methyllithium with the Doering–Moore–Skattebøl-type reaction (Scheme 17) [20,59], probably via a bracketed carbenoid intermediate. Carbene can be generated in aqueous medium with the Makosza method [60], in the presence of benzyltriethylammonium chloride in a mixture of aqueous 50% NaOH, trichloromethane, and hexane.
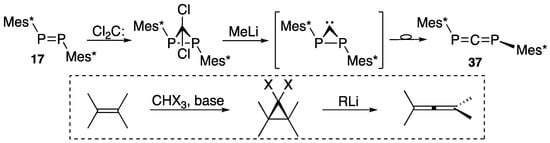
Scheme 17.
Preparation of diphosphaallene 37 with dichlorocarbene and the Doering–Moore–Skattebøl reaction.
This one-carbon homologation method [61] can also be applied to prepare phosphaallene (36), as shown in Scheme 18 with a combination of 33 and dichlorocarbene [59].
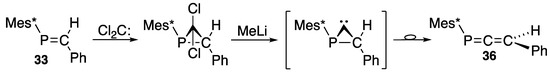
Scheme 18.
Preparation of phosphaallene 36 with dichlorocarbene.
Phosphacumulenes 36 and 37 have axial dissymmetry according to their X-ray structures [62,63,64], so, under achiral circumstances, the reaction products are expected to be a 1:1 mixture of enantiomers, though in Scheme 15, Scheme 16, Scheme 17 and Scheme 18 for 36 and 37, only the (S)-isomers were displayed. Actually both are mixtures of enantiomers and could be separated with high-performance liquid chromatography (HPLC). Each separated enantiomer is racemized on exposure to light due to the rotation around the axis of the molecules [47,65], but they were found to be thermally stable in the dark at room temperature, even at 50 °C for 15 h for 36.
As shown in Scheme 19 (third and fourth rows), the Doering–Moore–Skattebøl method can be applied to the P=C=C and P=C=P systems (41 and 37) to yield 1-phospha-1,2,3-butatriene 38 [49] and 1,4-diphospha-1,2,3-butatrienes (39 and 40, ca 4:1) [51], respectively, which implies that two carbon atoms are inserted into P=C and P=P in a stepwise manner. Additionally, Märkl described the preparation of extended cumulenes (38–40) from 2,4,6-tri-t-butylphenylphosphonous dichloride (15) and allenyllithium compounds as nucleophiles (Scheme 19, first and second rows) [50,52].
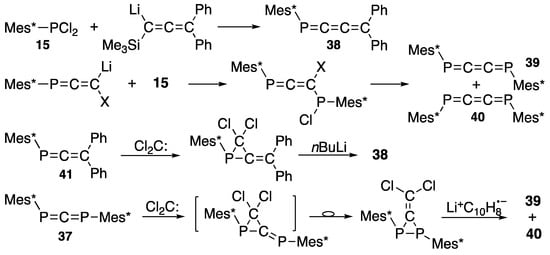
Scheme 19.
Preparation of phospha-1,2,3-butatrienes (38–40).
Table 3 also lists P=C-containing compounds such as 42 [53] and 43 [54], whose chemical shifts have higher values, which could be rationalized if their canonical forms are considered as depicted in Scheme 20, where the anion is located on the phosphorus atom.
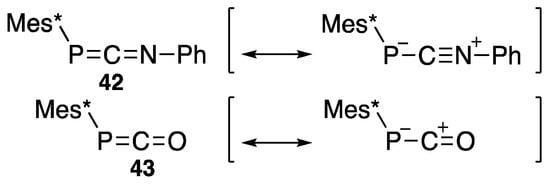
Scheme 20.
Canonical forms for oxygen- and nitrogen-incorporated phosphacumulenes.
3.3. Stable Phosphaalkynes
Phosphaalkynes have the lowest coordination number of 1, and once sterically protected, this type of compound can be isolated. Using tris(trimethylsilyl)phosphane (44) and acid chlorides, Becker [55] reported the t-butyl derivative 45 in 1981, as shown in Scheme 21. With a similar method, Märkl [56] described the 2,4,6-tri-t-butylphenyl derivative 46 in 1986.
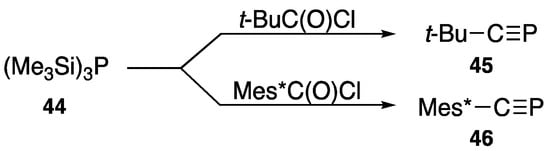
Scheme 21.
Preparation of phosphaalkynes (45 and 46).
Alternatively, phosphaethyne (46) can be prepared by an interesting rearrangement reaction, a phosphorus version of the Fritsch–Buttenberg–Wiechell rearrangement, as shown in Scheme 22 [66,67,68]. (E)-2-Chloro-1-(2,4,6-tri-t-butylphenyl)phosphaethene (47) is metalated to 48 with t-butyllithium at −78 °C, and the metalation is confirmed by methylation with iodomethane at that temperature to yield (E)-2-chloro-1-(2,4,6-tri-t-butylphenyl)-1-phosphapropene (49). However, after metalation, when 48 is allowed to warm up to room temperature, phosphaethyne 46 is obtained, indicating that the Mes* group migrates from phosphorus to carbon. On the other hand, if (Z)-2-chloro-1-(2,4,6-tri-t-butylphenyl)phosphaethene (50) is allowed to react with t-butyllithium, the obtained lithium compound (51) does not lead to phosphaethyne 46, while the formed alkylation product, (Z)-2-chloro-1-(2,4,6-tri-t-butylphenyl)-1-phosphapropene (52), indicates that the configuration is retained [67]. Based on a kinetic study of the Fritsch–Buttenberg–Wiechell rearrangement shown at the bottom of Scheme 22, Köbrich [69] suggested that phenyl group PhA migrates from one carbon to another, thus eliminating trans-halogen atom as halide to form acetylene [70].
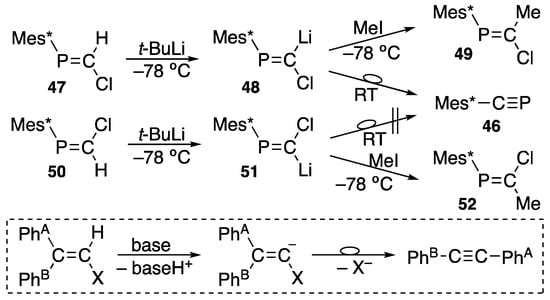
Scheme 22.
Preparation of phosphaalkyne 46 and the Fritsch–Buttenberg–Wiechell reaction; RT = room temperature.
In this rearrangement process from 47 to 46, a similar transition state such as 53 might be involved (Scheme 23). Appel [71] reported the formation of 46 from 2,2-dibromo-1-(2,4,6-tri-t-butylphenyl)phosphaethene (54), though it is not certain if phosphanylidenecarbenes such as 56 or 57 (Scheme 24) are involved in this formation of phosphaalkyne, whereas the carbene, if freely generated, is experimentally [72] and theoretically [73] known to yield an intramolecular insertion product such as 55.
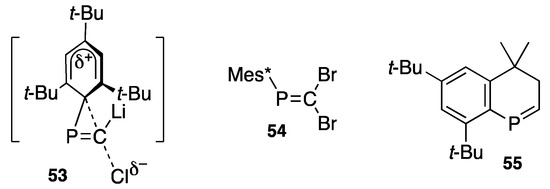
Scheme 23.
A plausible transition state (53) and compounds 54 and 55.
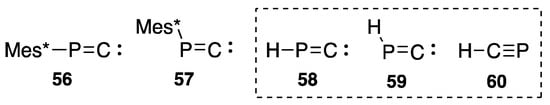
Scheme 24.
Structures of phosphanylidene carbenes and phosphaalkynes.
Concerning the energy difference between phosphanylidenecarbene (linear 56 or bent 57) and phosphaethyne 46, we calculated total energy with an ab initio method and found that 46 has total energy of 87.3 kcal/mol less than that of 56 or 57 [70]. Additionally, in 1986, Nguyen theoretically computed 83.9 kcal/mol as the difference between 58 and 60 in a model system [74], and Lehmann [75] calculated the difference between 59 and 60 as 84.1 kcal/mol in 1985. Each computed result indicates that phosphaalkynes are far more stable than the corresponding phosphanylidenecarbene isomers (Scheme 24).
3.4. Singlet Biradicals from Phosphaalkyne
An interesting reactivity of phosphaalkyne 46 (with the lowest coordination number of 1) is dimerization promoted by half of an equivalent of t-butyllithium to a singlet biradical species such as 1-t-butyl-3-methyl-2,4-bis(2,4,6-tri-t-butylphenyl)-1,3-diphosphacyclobutane-2,4-diyl (62) after quenching 61 with iodomethane (Scheme 25) [76,77]. In 1995, Niecke reported 2,4-dichloro-1,3-bis(2,4,6-tri-t-butylphenyl)-1,3-diphosphacyclobutane-2,4-diyl (64) [78] as a reductive coupling reaction product of 2,2-dichloro-1-(2,4,6-tri-t-butylphenyl)phosphaethene (63) at −100 °C. The radicals are on the carbon atoms in both cases, but the Mes* groups are contrarily located. An attempt to combine these two different singlet biradicals was described for a novel radical system [79].
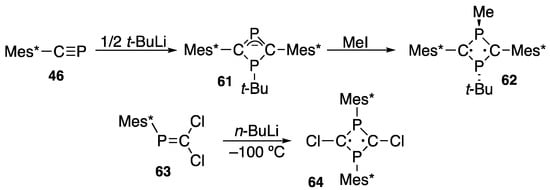
Scheme 25.
Two types of stable singlet biradicals (62 and 64).
Cyclic phosphide anion 61 serves as a good nucleophile to yield various kinds of biradical species in addition to methyl derivative 62. Anion 61 reacts with several arynes or benzynes to yield phenyl-substituted biradicals [80]. In place of the t-butyllithium shown in Scheme 25, a series of carbon nucleophiles (including t-butyl iodide and even nitrogen nucleophiles such as LDA (lithium diisopropylamide)) can be used [77]. It should be noted that nucleophiles initially attack the phosphorus atom to form 61, in contrast to the corresponding nitriles.
Compound 62 is a deep blue solid with an mp of 158–160 °C and thermally stable even in a solution in air. The 31P NMR spectrum shows an ABq at δP 55.9 (t-BuP) and −11.3 (MeP) ppm with 2JPP 362.8 Hz, and the 13C NMR spectrum of the ring C shows a dd at δC 111.3 (ring C) with 1JCP 10.7 and 3.3 Hz. An X-ray study indicated no apparently direct bonding between either P–P (2.43 Å) or C–C (2.50 Å) in the ring with two almost perfectly planar carbon atoms. No signal was observed in EPR measurements, indicating that the compound is a singlet biradical species. The chemical reactivity of singlet biradicals is of high interest, including hydrolysis with water [77], oxidation with molecular oxygen [77], TEMPO (2,2,6,6-tetramethyl-1-piperidinoxy) [77], and ammoniumyl antimonate [81] yielding a radical cation (66), sulfurization with elemental sulfur [82], reduction with hydride (67) [83,84], muonium addition [85], respectively. Oligomeric poly-biradical species 68 [86] can be prepared and pyrimidine derivatives such as 69 are promising for HF-capture reagents [87]. The formation of neutral radical 65, which is obtained via the partial oxidation of cyclic phosphide anion 61 with iodine, is also noteworthy. The radical is stable in air, and X-ray analyses, theoretical calculations, and EPR spectroscopic studies have indicated that 65 is a carbon-centered radical [88] (Scheme 26).
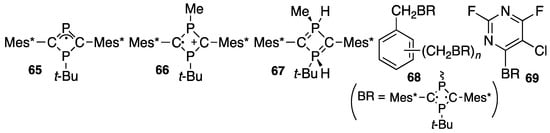
Scheme 26.
Some singlet biradical-related species.
4. Diphosphinidenecyclobutenes
4.1. Preparation of Diphosphinidenecyclobutenes
Diphosphinidenecyclobutenes, abbreviated as DPCBs, are prepared as depicted in Scheme 27. From 2,4,6-tri-t-butylphenylphosphane (16) or 2,4,6-tri-t-butylphenylphosphonous dichloride (15), (2-phenylethynyl)(2,4,6-tri-t-butylphenyl)phosphane (70) is prepared via chloro(2,4,6-tri-t-butylphenyl)phosphane. After metalation followed by oxidative coupling with 1,2-dibromoethane (DBE) at low temperature, the corresponding diphosphane 71 is formed, thus leading to 1,6-diphospha-1,2,4,5-hexatetraene 72 at room temperature through a Cope rearrangement. Upon heating, tetraene 72 rearranges to (E,E)-1,2-diphenyl-3,4-bis(2,4,6-tri-t-butylphenylphosphinidene)cyclobutene (73) [89,90]. Compound 73 was described by Appel in 1987 [91], and the corresponding 3,4-bis(trimethylsilyl) derivative (78 in Scheme 28) was prepared by ourselves with this phospha-Cope method [92]. Phosphinidenecyclobutene 73 can be prepared in other routes, e.g., as shown in the middle row of Scheme 27, from (2-phenylethynyl)(2,4,6-tri-t-butylphenyl)phosphinous chloride (74) followed by reductive coupling reaction that forms 71, whereas 74 is prepared from 2-phenylethynylphosphonous dichloride (75) and 2,4,6-tri-t-butylphenyllithium (14); as shown at the bottom of Scheme 27, from (1-phosphaallen-3-yl)lithium 76 followed by oxidative coupling reaction with DBE forms 72 [89,90]; and Scheme 15 suggests that, 1-phosphaallene 36, as a precursor of 76, can be prepared with the Peterson reaction of 2,2-dibromo-1-(2,4,6-tri-t-butylphenyl)phosphaethene (77) with benzaldehyde.
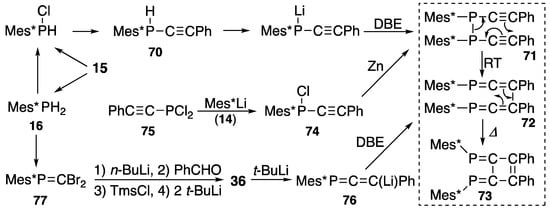
Scheme 27.
Preparation of diphosphinidenecyclobutene 73; DBE = 1,2-dibromoethane, RT = room temperature.
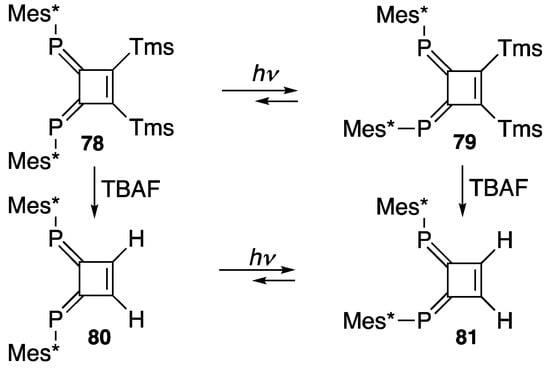
Scheme 28.
E/Z photoisomerization of 78 and 80; TBAF = n-Bu4N+F−.
As shown in Scheme 13 for phosphaethene 33, (E,E)-3,4-bis(2,4,6-tri-t-butylphenylphosphinidene)-1,2-bis(trimethylsilyl)cyclobutene (78) can undergo E/Z photoisomerization and yield (E,Z)-isomer 79 (Scheme 28). A simple DPCB 80, which is derived from 78 and tetrabutylammonium fluoride (TBAF), also shows a similar photoisomerization for 81. It should be noted that in both cases, even after a longer irradiation time, the (Z,Z)-isomer is not formed, probably due to the severe congestion around the adjacent Mes* groups [93].
It is noteworthy that iodine initiates E/Z isomerization, as shown in Scheme 29. (E,E)-DPCB 82 was found to be isomerized to the corresponding (E,Z)-isomer 83 in the presence of iodine in THF at room temperature for 1 h [94]. Facile rotation between phosphorus and carbon in cyclobutenylium iodide 84 could operate as an intermediate during the process of isomerization.
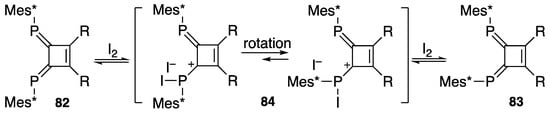
Scheme 29.
Isomerization from 82 to 83 induced by iodine; R = Ph, Tms, H.
Bulky groups such as Mes* on phosphorus atoms appear to suffer from free rotation around the P–C bond and thus hinder the rotation that distinguishes syn- and anti-rotamers for DPCB 85, which is protected with two unsymmetrical substituents such as 2,4-di-t-butyl-6-methylphenyl (Scheme 30). Two types of the tetracarbonyltungsten complexes of 85 were separated by column chromatography, and the syn-isomer could be analyzed by X-ray crystallography. The anti-isomer was further analyzed with a chiral HPLC column, and it was revealed that the isomer consists of two enantiomers, which was confirmed with the CD spectra [95].
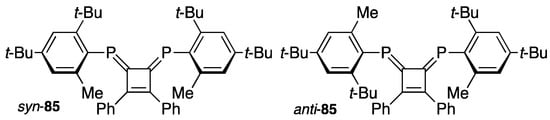
Scheme 30.
Rotational isomers syn- and anti-85.
As an interesting derivative related to DPCB, 1,2-diphospha[4]radialene 89 is prepared as shown in Scheme 31 from 1,2-dibenzyl-DPCB 87. (3-Phenylprop-1-yn-1-yl)(2,4,6-tri-t-butylphenyl)phosphane (86) is lithiated with phenyllithium, followed by oxidative coupling with 1,2-dibromoethane (DBE) to yield 1,2-dibenzyl DPCB 87, which is brominated with N-bromosuccinimide (NBS) to yield bis(α-bromobenzyl) derivative 88; finally, a reaction with butyllithium at −78 °C yields (E,E,E,E)-1,2-dibenzylidene-3,4-bis[(2,4,6-tri-t-butylphenyl)phosphinidene]cyclobutane (89). The configuration was confirmed with X-ray analysis; 89 appears to suffer from steric crowding between the two phenyl groups by taking the E-configuration, though they tilt about by 30° from the radialene plane in the same direction, while the Mes* groups are almost perpendicular to the plane so they avoid congestion within the molecule at least in the solid state [96].
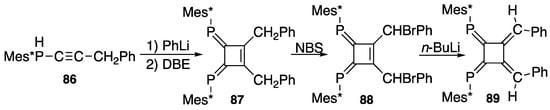
Scheme 31.
Preparation of diphospha[4}radialene 89; DBE = 1,2-dibromoethane, NBS = N-bromosuccinimide.
4.2. Transition Metal Complexes of Diphosphinidenecyclobutenes
As expected, DPCB 82 is a good bidentate ligand for transition metals such as chromium, molybdenum, tungsten, iron, gold, palladium, platinum, rhodium. Scheme 32 shows several typical examples (90–94) that have been analyzed in X-ray studies [97,98,99,100,101,102,103]. It is noteworthy that the molecular structure of 94 shows that the π-allyl system is perpendicular to the DPCB plane, which controls the stereochemistry in the catalytic reactions. The details of this phenomenon are described later in this section.
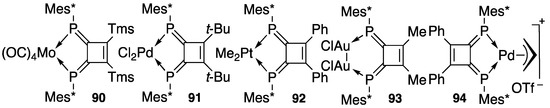
Scheme 32.
Some DPCB–metal complexes; Tf = CF3SO2.
The reaction of 1,2-diphenyl-3,4-bis(2,4,6-triisopropylphenylphosphinidene)cyclobutene (95) with pentacarbonyltungsten leads to an interesting result, as depicted in Scheme 33: it not only serves as a bidentate ligand to yield 97 but also it yields 96 as a monodentate ligand. More interestingly, the ligand serves as a side-on ligand to yield 98 [104].
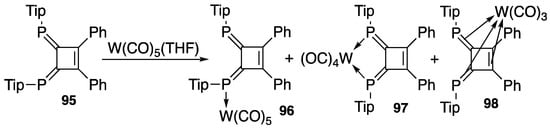
Scheme 33.
Tungsten carbonyl complexes of 95; Tip = 2,4,6-i-Pr3C6H2.
4.3. Synthetic Application to Catalytic Reactions of DPCB–Transition Metal Complexes
DPCB complexes are effective catalysts for organic reactions [36,103,105,106,107]. Scheme 34 demonstrates some of the cross-coupling reactions—such as the Sonogashira [99] (A), Suzuki–Miyaura (B) [105], and Migita–Kosugi–Stille (C) [108] reactions catalyzed by DPCB–transition metal complexes—with representative examples.
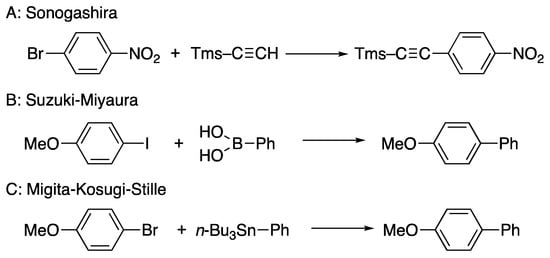
Scheme 34.
Some cross-coupling reactions catalyzed by DPCB–metal complexes. (A) Sonogashira reaction, (B) Suzuki–Miyaura reaction, (C) Migita–Kosugi–Stille reaction.
Scheme 35 shows some other examples. The Ullmann-type amination reactions (A) can be executed with a DPCB catalyst without a solvent at room temperature [109,110]. Hydroamination to 1,3-dienes (B) can be induced at room temperature without a solvent [103]. An enyne metathesis reaction (C) proceeds via a DPCB–gold complex 93 [101]. Cyanation reactions (D) are widely applied to convert halobenzenes to cyanobenzenes catalyzed by 94 [111]. Ethylene polymerization (E) is catalyzed by thermally stable palladium and a platinum DPCB-ligated catalyst such as 91 and 92 [97,100].
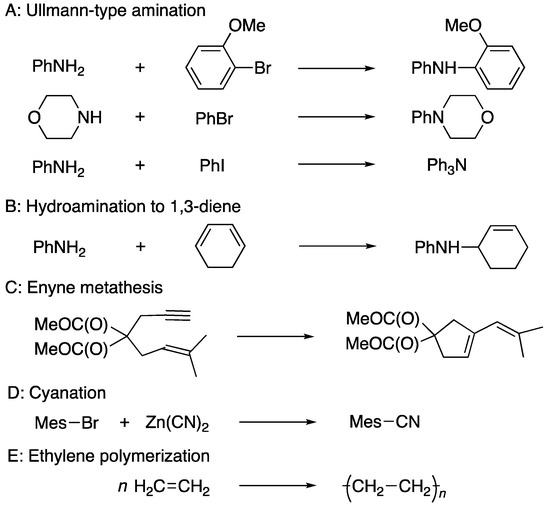
Scheme 35.
Ullmann-type amination (A), hydroamination to 1,3-diene (B), enyne metathesis (C), cyanation (D), and ethylene polymerization (E) catalyzed by DPCB–metal complexes. Mes = 2,4,6-Me3C6H2.
A stable (π-allyl)palladium complex such as 94 (shown in Scheme 32) [103] has shown a wide range of catalytic reactivity including in the Tsuji–Trost-type reaction that enables allylic alcohols to yield amines via the elimination of water [36,105,106] under mild conditions such as room temperature for 1 h with a 0.1 mol% DPCB–palladium catalyst.
Scheme 36 [112] shows that, starting from either from primary or secondary C4-allylic alcohol (99 or 100), products 101 and 102 are regioselectively obtained in the same ratio (86:14) and the same yield (95%), indicating that (π-allyl)palladium complex 103 is a common intermediate.
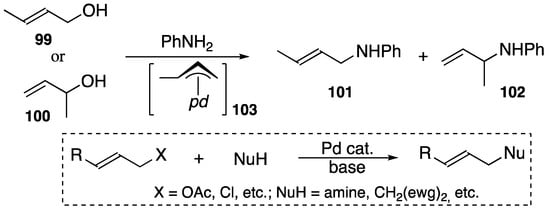
Scheme 36.
DPCB–palladium-catalyzed amination from C4-allylic alcohols and the Tsuji–Trost reaction; ewg = electron-withdrawing group, pd = (DPCB)Pd+.
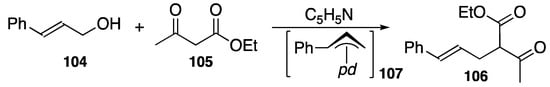
Direct alkylation can be regioselectively achieved using carbanions from active methylene compounds such as ethyl acetylacetate (105) as a nucleophile in order to formally substitute the OH group of allylic alcohol 104 with an alkylation product (106) in a high yield, as shown in Scheme 37 [112], where an intermediate 107 is supposed to be functional. It is not necessary to convert alcohol 104 to a more reactive derivative (such as an ester or halide) as an allylic starting material in advance of conventional Tsuji–Trost reactions.
Scheme 37.
DPCB–palladium-catalyzed Tsuji–Trost-type alkylation reaction; pd = (DPCB)Pd+.
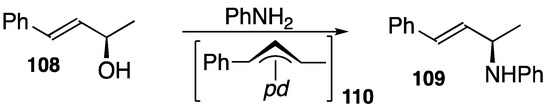
Furthermore, the reaction from a chiral alcohol 108 of 98.5 %ee yields corresponding amine 109 of the same chirality in a 92% yield, which indicates that a double inversion process is involved and surrounds intermediate 110 during the reaction course to control the stereochemistry (Scheme 38) [112].
Scheme 38.
DPCB–palladium-catalyzed amination of chiral alcohol to amine; pd = (DPCB)Pd+.
These results indicate that DPCB ligands have a high coordination ability due to their σ-donation/π-back donation interactions, strong π-acceptor property due to their low-lying LUMO (π*), and unique coordination structure with planarity. In addition, the catalysts are highly stable toward air and reusable after reactions in most cases. The above characteristics are superior to catalysts with ligands having sp3 phosphorus or sp2 nitrogen atoms for catalytic organic reactions.
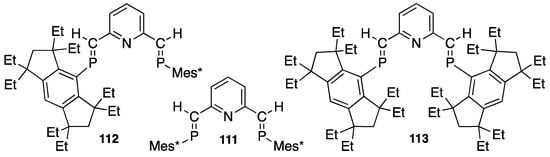
Recently, Ozawa reported using pincer ligands such as 111–113 (Scheme 39) to activate CO bonds in carbon dioxide, taking advantage of the frustrated lone pair (FLP) within the ligand [113,114]. Further fundamental and applied investigation are expected to lead to progress in research of this type of low-coordinate phosphorus ligand for ideally efficient catalytic reactions.
Scheme 39.
Some pincer ligands with sterically bulky P=C bonds (111–113).
5. Closing Remarks
Forty years have passed since a true “phosphobenzene” was prepared for the first time in 1981 via the steric protection methodology with an extremely bulky 2,4,6-tri-t-butylphenyl group (Mes*). It was believed, even theoretically, that molecules such as those with heavier main-group elements never existed as stable compounds, but various kinds of “unusual” phosphorus compounds have been isolated and characterized by utilizing the Mes* group. Interest in basic research on this class of compounds and synthetic applications for organic synthesis is high. This review article was based on a plenary lecture delivered by the author at the 23rd International Conference on Phosphorus Chemistry (ICPC-23) remotely held in Częstochowa, Poland, 5–9 July 2021 [115].
Funding
This research received no external funding.
Acknowledgments
The author notes the University of Tokyo and Tohoku University (Sendai) as his affiliated institutions. In addition, he thanks Reinhard Schmutzler and Wolf-Walther du Mont (Braunschweig, Germany), Edgar Niecke and Rainer Streubel (Bonn, Germany), and Anthony J. Arduengo, III (Tuscaloosa, U.S.A.) for inviting him to conduct his research on phosphorus chemistry at their universities. He also thanks all the collaborators whose names appear in the references for their dedicated research activities and interest in this science. Financial support in part by Japan Society for the Promotion of Science, National Science Foundation (U.S.A.), and Alexander von Humboldt Foundation (Germany) is gratefully acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References
- Okazaki, R.; Hosogai, T.; Iwadare, E.; Hashimoto, M.; Inamoto, N. Preparation of sterically hindered nitrosobenzenes. Bull. Chem. Soc. Jpn. 1969, 42, 3611–3612. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.G. Syntheses and reactions of some hindered organophosphorus compounds. J. Org. Chem. 1965, 30, 1262–1263. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Fujishima, I.; Okazaki, R.; Inamoto, N. Reaction of 1,3,5-tri-tert-butylbenzene with phosphorus trichloride: Revised structure of 2,4,6-tri-tert-butylphenylphosphinic chloride. Chem. Ind. (Lond.) 1970, 19, 625. [Google Scholar]
- Köhler, H.; Michaelis, A. Über Phenylphosphin und Phosphobenzol (Diphosphenyl). Ber. Dtsch. Chem. Ges. 1877, 10, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Kuchen, W.; Buchwald, H. Zur Kenntnis der Organophosphorverbindungen, 1. Das Tetraphenyl-cyclotetraphosphin. Chem. Ber. 1958, 91, 2296–2304. [Google Scholar] [CrossRef]
- Daly, J.J.; Maier, L. Molecular structure of phosphobenzene. Nature 1964, 203, 1167–1168. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of phosphobenezene A, (PC6H5)5. J. Chem. Soc. 1964, 6147–6166. [Google Scholar] [CrossRef]
- Daly, J.J.; Maier, L. Structure of phosphobenzene. Nature 1965, 208, 383–384. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of a trigonal form of phosphobenezene B, (PC6H5)6. J. Chem. Soc. 1965, 4789–4799. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of a triclinic form of phosphobenzene, (PC6H5)6, referred as B. J. Chem. Soc. A 1966, 428–439. [Google Scholar] [CrossRef]
- Yoshifuji, M. 2,4,6-Tri-tert.-butylphenylphosphane, (E)-1,2-bis(2,4,6-tri-tert.-butylphenyl)diphosphene, (E)-1-(2,4,6-tri-methylphenyl)-2-(2,4,6-tri-tert.-butylphenyl)diphosphene. In Synthetic Methods in Inorganic and Organometallic Chemistry: Phosphorus, Arsenic, Antimony, and Bismuth; Herrmann, W.A., Karsch, H.H., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1996; Volume 3, pp. 118–125. [Google Scholar]
- Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene; Isolation of a true “phosphobenzene”. J. Am. Chem. Soc. 1981, 103, 4587–4589. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene; Isolation of a true "phosphobenzene" (correction). J. Am. Chem. Soc. 1982, 104, 6167. [Google Scholar] [CrossRef]
- Hamaguchi, H.; Tasumi, M.; Yoshifuji, M.; Inamoto, N. The P=P stretching frequency observed in the resonance Raman spectrum of bis(2,4,6-tri-tert-butylphenyl)diphosphene. J. Am. Chem. Soc. 1984, 106, 508–509. [Google Scholar] [CrossRef]
- Cetinkaya, B.; Hudson, A.; Lappert, M.F.; Goldwhite, H. Generation and e.s.r. spectra of some new phosphorus-centred radicals Ṗ2Ar2X, Ṗ(Ar)X, Ṗ(OAr)2, ṖAr2(:O), ṖAr[N(SiMe3)2](:NSiMe3), and [P2Ar2]˙– derived from the bulky group C6H2But3-2,4,6 (= Ar). J. Chem. Soc. Chem. Commun. 1982, 609–610. [Google Scholar] [CrossRef]
- Cowley, A.H.; Kilduff, J.E.; Newman, T.H.; Pakulski, M. Diphosphenes (RP:PR). Synthesis and NMR characterization. J. Am. Chem. Soc. 1982, 104, 5820–5821. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Watanabe, T. Reduction of diphosphene; Formation of dl- and meso-diphosphanes. Chem. Lett. 1983, 12, 585–588. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Matsushita, T.; Nishimoto, K. Isolation of some sterically protected unsymmetrical diphosphenes; Nature of the phosphorus-phosphorus double bond. J. Am. Chem. Soc. 1983, 105, 2495–2497. [Google Scholar] [CrossRef]
- Yoshifuji, M. 31P NMR in studies of some one- and two-coordinate phosphorus compounds. In Phosphorus-31 NMR Spectral Properties in Compound Characterization and Structural Analysis; Quin, L.D., Verkade, J., Eds.; VCH Publishers: New York, NY, USA, 1994; pp. 175–187. [Google Scholar]
- Yoshifuji, M.; Sasaki, S.; Inamoto, N. The first preparation of an unsymmetrical 1,3-diphospha-allene carrying 2,4,6-tri-t-butylphenyl and 2,4,6-tri-t-pentylphenyl as protecting groups. J. Chem. Soc. Chem. Commun. 1989, 22, 1732–1733. [Google Scholar] [CrossRef]
- Smit, C.N.; van der Knaap, T.A.; Bickelhaupt, F. Stable unsymmetrical diphosphenes. Tetrahedron Lett. 1983, 24, 2031–2034. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Inamoto, N. Reaction of a diaryldiphosphene with hexacarbonylchromium(0)–Formation of (arene)tricarbonylchromium(0) complexes. Tetrahedron Lett. 1983, 24, 4855–4858. [Google Scholar] [CrossRef]
- Cowley, A.H.; Kilduff, J.E.; Mehrotra, S.K.; Norman, N.C.; Pakulski, M. A new approach to the formation of phosphorus–phosphorus double bonds. J. Chem. Soc. Chem. Commun. 1983, 9, 528–529. [Google Scholar] [CrossRef]
- Jutzi, P.; Meyer, U.; Krebs, B.; Dartmann, M. Bis(pentamethylcyclopentadienyl)diphosphene—A molecule with fluxional structure and reactive P≡C bond. Angew. Chem. Int. Ed. Engl. 1986, 25, 919–921. [Google Scholar] [CrossRef]
- Couret, C.; Escudié, J.; Satgé, J. Un nouveau diphosphene stable: Le bis[tris(trimethylsilyl)methyl]diphosphene. Tetrahedron Lett. 1982, 23, 4941–4942. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Sato, T.; Inamoto, N. Wavelength- and temperature-dependent photolysis of a diphosphene. Generation of 2,4,6-tri-t-butylphenylphosphinidene and E/Z isomerization. Chem. Lett. 1988, 17, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Caminade, A.-M.; Verrier, M.; Ades, C.; Paillous, N.; Koenig, M. Laser irradiation of a diphosphene: Evidence for the first cis–trans isomerization. J. Chem. Soc. Chem. Commun. 1984, 13, 875–877. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shinohara, N.; Toyota, K. Application of 1,2-bis(2-bromo-3,5-di-t-butylphenyl)ethane to preparation of compounds having two diphosphene units. Tetrahedron Lett. 1996, 37, 7815–7818. [Google Scholar] [CrossRef]
- Smith, R.C.; Shah, S.; Protasiewicz, J.D. A role for free phosphinidenes in the reaction of magnesium and sterically encumbered ArPCl2 in solution at room temperature. J. Organomet. Chem. 2002, 646, 255–261. [Google Scholar] [CrossRef]
- West, R.; Fink, M.J.; Michl, J. Tetramesityldisilene, a stable compound containing a silicon-silicon double bond. Science 1981, 214, 1343–1344. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected organophosphorus compounds of unusual structures. Pure Appl. Chem. 2017, 89, 281–286. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected organophosphorus compounds of unusual structures. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1452–1456. [Google Scholar] [CrossRef]
- Yoshifuji, M. Phosphinylidenephosphines (diphosphenes). In Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, M., Scherer, O.J., Eds.; Thieme: Stuttgart, Germany, 1990; pp. 321–337. [Google Scholar]
- Weber, L. The chemistry of diphosphenes and their heavy congeners: Synthesis, structure, and reactivity. Chem. Rev. 1992, 92, 1839–1905. [Google Scholar] [CrossRef]
- Dillon, K.B.; Mathey, F.; Nixon, J.F. Phosphorus: The Carbon Copy from Organophosphorus to Phospha-Organic Chemistry; John Wiley & Sons: Chichester, UK, 1998. [Google Scholar]
- Yoshifuji, M. Recent developments in the chemistry of low-coordinated organophosphorus compounds. Pure Appl. Chem. 2005, 77, 2011–2020. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected diphosphenes. Eur. J. Inorg. Chem. 2016, 5, 607–615. [Google Scholar] [CrossRef]
- Yoshifuji, M. Protecting groups for stabilization of unusual organophosphorus compounds. Main Group Chem. News 1998, 6, 20–26. [Google Scholar]
- Yoshifuji, M. Protecting groups for stabilization of inter-element linkages. J. Organomet. Chem. 2000, 611, 210–216. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Reaction of the diphosphene ArP=PAr (Ar=2,4,6-But3C6H2) with sulphur: Isolation and X-ray structure of the diphosphene monosulphide. J. Chem. Soc. Chem. Commun. 1983, 862–863. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ando, K.; Shibayama, K.; Inamoto, N.; Hirotsu, K.; Higuchi, T. The first X-ray structure analysis of a thiadiphosphirane. Angew. Chem. Int. Ed. Engl. 1983, 22, 418–419. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Hashida, T.; Inamoto, N.; Hirotsu, K.; Horiuchi, T.; Higuchi, T.; Ito, K.; Nagase, S. E/Z Photoisomerization of a diphosphene of carbonyl metal complexes (M = Cr, Mo, W). Angew. Chem. Int. Ed. Engl. 1985, 24, 211–212. [Google Scholar] [CrossRef]
- Märkl, G. 2,4,6-Triphenylphosphabenzene. Angew. Chem. Int. Ed. 1966, 5, 846–847. [Google Scholar] [CrossRef]
- Klebach, T.C.; Lourens, R.; Bickelhaupt, F. Synthesis of mesityldiphenylmethylenephosphine: A stable compound with a localized phosphorus:carbon bond. J. Am. Chem. Soc. 1978, 100, 4886–4888. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Inamoto, N. Photoisomerization of benzylidenephosphine containing phosphorus in low coordination state. Tetrahedron Lett. 1985, 26, 1727–1730. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Matsuda, I.; Niitsu, T.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structures of E- and Z-phosphaethylenes. Tetrahedron 1988, 44, 1363–1367. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Okamoto, Y.; Asakura, T. Separation of an optically active phosphaallene of pseudo axial dissymmetry. Tetrahedron Lett. 1990, 31, 2311–2314. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Inamoto, N. Isolation and characterisation of the first stable diphospha-allene; P,P’-bis(2,4,6-tri-t-butylphenyl)-1,3-diphospha-allene. J. Chem. Soc. Chem. Commun. 1984, 689–690. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Yoshimura, H.; Hirotsu, K.; Okamoto, A. Reactions of dichlorocarbene with sterically protected phosphaallene and 1,3-diphosphaallene. J. Chem. Soc. Chem. Commun. 1991, 2, 124–125. [Google Scholar] [CrossRef]
- Märkl, G.; Sejpka, H.; Dietl, S.; Nuber, B.; Ziegler, M.L. 1-Phospha-1,2,3-butatriene. Angew. Chem. Int. Ed. 1986, 25, 1003–1004. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Yoshimura, H. Preparation of 1,4-diphosphabutatriene from 3-dichloromethylene-1,2-diphosphirane. Chem. Lett. 1991, 20, 491–492. [Google Scholar] [CrossRef]
- Märkl, G.; Kreitmeier, P. 1,4-Diphosphabutatriene. Angew. Chem. Int. Ed. 1988, 27, 1360–1361. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Shibayama, K.; Inamoto, N. Isolation and characterization of some very stable 1-phospha-allenes; sterically protected iminomethylene- and ethenylidenephosphines. Tetrahedron Lett. 1984, 25, 1809–1812. [Google Scholar] [CrossRef]
- Appel, R.; Paulen, W. The first stable phosphaketene. Angew. Chem. Int. Ed. Engl. 1983, 22, 785–786. [Google Scholar]
- Becker, G.; Gresser, G.; Uhl, W. Acyl- und Alkylidenphosphane, XV. 2,2-Dimethylpropylidinphosphan, eine stabile Verbindung mit einem Phosphoratom der Koordinationszahl 1. Z. Naturforsch. B 1981, 36, 16–19. [Google Scholar] [CrossRef]
- Märkl, G.; Sejpka, H. 2-(2,4,6-Tri-tert-butylphenyl)-1-phosphaethin, 1,4-bis-(trimethylsiloxy)-1,4-bis(2,4,6-tri-tert-butylphenyl)-2,3-diphosphabutadien. Tetrahedron Lett. 1986, 27, 171–174. [Google Scholar] [CrossRef]
- Appel, R.; Casser, C.; Immenkeppel, M.; Knoch, F. Easy synthesis of phosphaalkenes by a phosphorus-analogous isocyanide reaction and an atypical crystal structure of a tetracarbonyl(phosphaalkene)iron complex. Angew. Chem. Int. Ed. Engl. 1984, 23, 895–896. [Google Scholar] [CrossRef]
- Karsch, H.H.; Köhler, F.H.; Reisacher, H.-U. C-Phosphinosubstituierte Phosphaalkene und eine erstes Carbodiphosphan. Tetrahedron Lett. 1984, 25, 3687–3690. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Yoshimura, H.; Toyota, K. Reaction of dichlorocarbene with phosphaethylenes. Preparation of phosphaallene from phosphaethylene via dichlorophosphirane. Chem. Lett. 1990, 19, 827–830. [Google Scholar] [CrossRef]
- Makosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Sasaki, S.; Niitsu, T.; Inamoto, N. A convenient new route from diphosphene to 1,3-diphospha-allene and dynamic NMR studies of the 2,4,6-tri-t-butylphenyl derivative. Tetrahedron Lett. 1989, 30, 187–188. [Google Scholar] [CrossRef]
- Karsch, H.H.; Reisacher, H.-U.; Müller, G. Molecular structure of a 1,3-diphosphaallene: (2,4,6-tBu3C6H2)P=C=P(2,4,6-tBu3C6H2), a phosphorus analogue of carbon disulfide. Angew. Chem. Int. Ed. 1984, 23, 618–619. [Google Scholar] [CrossRef]
- Ito, S.; Sekiguchi, S.; Yoshifuji, M. Formation and electrochemical properties of a 1,3-diphosphafulvene including formal dimerization of 1-phosphaallene. J. Org. Chem. 2004, 69, 4181–4184. [Google Scholar] [CrossRef]
- Ito, S.; Sekiguchi, S.; Freytag, M.; Yoshifuji, M. An efficient synthetic method of 1-(2,4,6-tri-t-butylphenyl)-3-phenyl-1-phosphaallenes [Mes*P=C=CR–Ph; R = H, SiMe3] from dibromophosphaethene [Mes*P=CBr2] (Mes* = 2,4,6-t-Bu3C6H2). Bull. Chem. Soc. Jpn. 2005, 78, 1142–1144. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Niitsu, T.; Inamoto, N.; Okamoto, Y.; Aburatani, R. The first separation of an optically active 1,3-diphospha-allene of axial dissymmetry. J. Chem. Soc. Chem. Commun. 1986, 20, 1550–1551. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Niitsu, T.; Inamoto, N. Formation of a phospha-alkyne via migration of the phenyl group from phosphorus to carbon. Chem. Lett. 1988, 17, 1733–1734. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Chemistry of phosphanylidene carbenoids. Top. Curr. Chem. 2003, 223, 67–89. [Google Scholar]
- Yoshifuji, M.; Kawanami, H.; Kawai, Y.; Toyota, K.; Yasunami, M.; Niitsu, T.; Inamoto, N. A new preparative route to phospha ethynes from dichlorophosphaethenes by lithiation, photoisomerization, and re-lithiation involving migration. Chem. Lett. 1992, 21, 1053–1056. [Google Scholar] [CrossRef]
- Köbrich, G.; Akhtar, A.; Ansari, F.; Breckoff, W.E.; Büttner, H.; Drischel, W.; Fischer, R.H.; Flory, K.; Fröhlich, H.; Goyert, W.; et al. Chemistry of stable α-halogenoorganolithium compounds and the mechanism of carbenoid reactions. Angew. Chem. Int. Ed. Engl. 1967, 6, 41–52. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Phosphorus version of the Fritsch-Buttenberg-Wiechell reaction. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 140–145. [Google Scholar] [CrossRef]
- Appel, R.; Immenkeppel, M. Niederkoordinierte Phosphorverbindungen, 58. Synthese und Reaktivität der 2,4,6-Tri-tert-butylphenyl(halogenmethylen)phosphane. Z. Anorg. Allg. Chem. 1987, 553, 7–14. [Google Scholar] [CrossRef]
- Ito, S.; Toyota, K.; Yoshifuji, M. Formation of 5,7-di-tert-butyl-1,1-dimethyl-4-phospha-1,2-dihydronaphthalene by the reaction of 2,2-dibromo-1-(2,4,6-tri-tert-butylphenyl)-1-phosphaethene with butyllithium. Chem. Commun. 1997, 1637–1638. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Theoretical studies of the intramolecular cyclization of 2,4,6-tBu3C6H2P=C: And effects of conjugation between the P=C and aromatic moieties. Beilstein J. Org. Chem. 2014, 10, 1032–1036. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Ha, T.K. Ab initio study of structures and relative stabilities of RCP (R = H, F) and their energetically higher-lying isomers RPC. J. Mol. Struct. (Theochem.) 1986, 139, 145–152. [Google Scholar] [CrossRef]
- Lehmann, K.K.; Ross, S.C.; Lohr, L.L. Experimental and ab initio determination of the bending potential of HCP. J. Chem. Phys. 1985, 82, 4460–4469. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, H.; Ito, S.; Yoshifuji, M. Synthesis of a 1,3-diphosphacyclobutane-2,4-diyl from Mes*C P (Mes* = 2,4,6-tBu3C6H2). Angew. Chem. Int. Ed. 2003, 42, 3802–3804. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Ito, S.; Yoshifuji, M. Preparation and reactions of 1,3-diphosphacyclobutane-2,4-diyls featuring an amino substituent and/or a carbonyl group. Chem. Eur. J. 2004, 10, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Niecke, E.; Fuchs, A.; Baumeister, F.; Nieger, M.; Schoeller, W.W. A P2C2-four-membered ring with unusual bonding–synthesis, structure, and ring opening of a 1,3-diphosphacyclobutane-2,4-diyl. Angew. Chem. Int. Ed. 1995, 34, 555–557. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Hirano, Y.; Schnakenburg, G.; Streubel, R.; Niecke, E.; Ito, S. New aspects of 1,3-diphosphacyclobutane-2,4-diyls. Helv. Chim. Acta 2012, 95, 1723–1729. [Google Scholar] [CrossRef]
- Ueta, Y.; Mikami, K.; Ito, S. Access to air-stable 1,3-diphosphacyclobutane-2,4-diyls by an arylation reaction with arynes. Angew. Chem. Int. Ed. 2016, 55, 7525–7529. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Arduengo, A.J., III; Konovalova, T.A.; Kispert, L.D.; Kikuchi, M.; Ito, S. Oxidation of 1,3-diphosphacyclobutane-2,4-diyl with ammoniumyl antimonate and EPR study of the corresponding cation radical. Chem. Lett. 2006, 35, 1136–1137. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, M.; Sugiyama, H.; Yoshifuji, M. Synthesis and properties of air-stable 1,3-diphosphacyclobutane-2,4-diyls and the related compounds. J. Organomet. Chem. 2007, 692, 2761–2767. [Google Scholar] [CrossRef]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Modeling the direct activation of dihydrogen by a P2C2-cyclic biradical: Formation of a cyclic bis(P-H λ5-phosphorane). Inorg. Chem. 2009, 48, 8063–8065. [Google Scholar] [CrossRef]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Diverse reactions of sterically protected 1,3-diphosphacyclobutane-2,4-diyls with hydride. Dalton Trans. 2010, 39, 8281–8287. [Google Scholar] [CrossRef]
- Ito, S.; Ueta, Y.; Koshino, K.; Kojima, K.M.; McKenzie, I.; Mikami, K. Observation of a metastable P-heterocyclic radical by muonium addition to a 1,3-diphosphacyclobutane-2,4-diyl. Angew. Chem. Int. Ed. 2018, 57, 8608–8613. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Poly(biradicals): Oligomers of 1,3-diphosphacyclobutane-2,4-diyl units. Angew. Chem. Int. Ed. 2008, 47, 6418–6421. [Google Scholar] [CrossRef] [PubMed]
- Ueta, Y.; Mikami, K.; Ito, S. Chemical detection of hydrogen fluoride by the phosphorus congener of cyclobutane-1,3-diyl. Inorg. Chem. 2015, 54, 8778–8785. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, M.; Yoshifuji, M.; Arduengo, A.J., III; Konovalova, T.A.; Kispert, L.D. Preparation and characterization of an air-tolerant 1,3-diphosphacyclobuten-4-yl radical. Angew. Chem. Int. Ed. 2006, 45, 4341–4345. [Google Scholar] [CrossRef] [PubMed]
- Yoshifuji, M. Chemistry of diphosphinidenecyclobutenes. J. Synth. Org. Chem. Jpn. 2003, 61, 1116–1123. [Google Scholar] [CrossRef]
- Toyota, K.; Horikawa, K.; Jensen, R.S.; Omori, K.; Kawasaki, S.; Ito, S.; Yoshifuji, M.; Morita, N. Improved preparative method for 1,2-diaryl-3,4-diphosphinidenecyclobutenes and its application to the studies of 1,2-bis(arylthienyl)-3,4-diphosphinidenecyclobutenes. Bull. Chem. Soc. Jpn. 2007, 80, 1580–1586. [Google Scholar] [CrossRef]
- Appel, R.; Winkhaus, V.; Knoch, F. Niederkoordinierte Phosphorverbindungen, 56. Valenzisomerisierung eines 3,4-Diphospha-1,5-hexadiins zu einem 3,4-Bis(phosphamethylen)-1-cyclobuten. Chem. Ber. 1987, 120, 243–245. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Murayama, M.; Yoshimura, H.; Okamoto, A.; Hirotsu, K.; Nagase, S. Preparation of sterically protected 3,4-bis(phosphinidene)cyclobutenes and their isomerism. Chem. Lett. 1990, 2195–2198. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M.; Nagase, S. Preparation and isomerization of 3,4-bis(2,4,6-tri-t-butylphenylphosphinidene)cyclobutenes. Bull. Chem. Soc. Jpn. 1992, 65, 2297–2299. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Abe, T.; Yoshifuji, M. Iodine-induced E/Z isomerization of sterically protected 3,4-diphosphinidenecyclobutenes and 1-phosphaethenes. Heteroat. Chem. 1994, 5, 549–553. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ichikawa, Y.; Toyota, K.; Kasashima, E.; Okamoto, Y. Rotational isomers of a 1,2-diphenyl-3,4-diphosphinidenecyclobutene. Chem. Lett. 1997, 26, 87–88. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M. Synthesis and structure of diphospha[4]radialene. Angew. Chem. Int. Ed. Engl. 1993, 32, 1163–1165. [Google Scholar] [CrossRef]
- Ozawa, F.; Kawagishi, S.; Ishiyama, T.; Yoshifuji, M. Synthesis, Structures, and Reactions of Methylplatinum(II) and -palladium(II) Complexes Bearing Diphosphinidenecyclobutene Ligands (DPCB-Y). Organometallics 2004, 23, 1325–1332. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M.; Miyahara, I.; Hayashi, A.; Hirotsu, K. X-Ray structure analyses of a diphosphinidenecyclobutene and its chelate type tetracarbonylmolybdenum(0) complex. J. Organometal. Chem. 1992, 431, C35–C41. [Google Scholar] [CrossRef]
- Toyota, K.; Masaki, K.; Abe, T.; Yoshifuji, M. Preparation and structure of dichloropalladium(II) complexes of sterically protected diphosphinidenecyclobutene and their synthetic application as homogeneous catalyst. Chem. Lett. 1995, 24, 221–222. [Google Scholar] [CrossRef]
- Ikeda, S.; Ohhata, F.; Miyoshi, M.; Tanaka, R.; Minami, T.; Ozawa, F.; Yoshifuji, M. Synthesis and reactions of palladium and platinum complexes bearing diphosphinidenecyclobutene ligands: A thermally stable catalyst for ethylene polymerization. Angew. Chem. Int. Ed. 2000, 39, 4512–4513. [Google Scholar] [CrossRef]
- Freytag, M.; Ito, S.; Yoshifuji, M. Coordination behavior of sterically protected phosphaalkenes on the AuCl moietyleading to catalytic 1,6-enyne cycloisomerization. Chem. Asian J. 2006, 1, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Freytag, M.; Yoshifuji, M. Structural and coordination properties of 1,2-bis(cyclopropyl)-3,4-bis(tri-t-butylphenyl)-3,4-diphosphinidenecyclobutene prepared by dehydrogenative homocoupling of 3-cyclopropyl-1-phosphaallene. J. Chem. Soc. Dalton 2006, 5, 710–713. [Google Scholar] [CrossRef]
- Minami, T.; Okamoto, H.; Ikeda, S.; Tanaka, R.; Ozawa, F.; Yoshifuji, M. (π-Allyl)palladium complexes bearing diphosphinidenecyclobutene ligands: A highly active catalyst for hydroamination of 1,3-dienes. Angew. Chem. Int. Ed. 2001, 40, 4501–4503. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ichikawa, Y.; Yamada, N.; Toyota, K. Preparation and X-ray analysis of novel carbonyltungsten(0) complexes of diphosphinidenecyclobutenes. Chem. Commun. 1998, 1, 27–28. [Google Scholar] [CrossRef]
- Ozawa, F.; Yoshifuji, M. Catalytic applications of transition metal complexes bearing diphosphinidenecyclobutenes (DPCB). Dalton Trans. 2006, 42, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Yoshifuji, M. Synthesis and catalytic properties of diphosphinidenecyclobutene-coordinated palladium and platinum complexes. C. R. Chim. 2004, 7, 747–754. [Google Scholar] [CrossRef]
- Ozawa, F.; Katayama, H.; Toyota, K.; Yoshifuji, M. Catalytic applications of transition metal complexes bearing sp2 hybridized phosphorus compounds. Catal. Catal. 2005, 47, 544–549. [Google Scholar]
- Gajare, A.S.; Jensen, R.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Low coordinated diphosphinidene ligands: A new entry for Stille cross-coupling of aryl bromides. Synlett 2005, 144–148. [Google Scholar] [CrossRef]
- Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Solvent free amination reactions of aryl bromides at room temperature catalyzed by a (π-allyl)palladium complex bearing a diphosphinidenecyclobutene ligand. J. Org. Chem. 2004, 69, 6504–6506. [Google Scholar] [CrossRef]
- Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Application of a diphosphinidenecyclobutene ligand in solvent free Cu-catalysed amination reactions of aryl halides. Chem. Commun. 2004, 17, 1994–1995. [Google Scholar] [CrossRef]
- Jensen, R.S.; Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. A convenient procedure for palladium catalyzed cyanation using a unique bidentate phosphorus ligand. Tetrahedron Lett. 2005, 46, 8645–8647. [Google Scholar] [CrossRef]
- Ozawa, F.; Okamoto, H.; Kawagishi, S.; Yamamoto, S.; Minami, T.; Yoshifuji, M. (π-Allyl)palladium complexes bearing diphosphinidenecyclobutene ligands (DPCB): Highly active catalysts for direct conversion of allylic alcohols. J. Am. Chem. Soc. 2002, 124, 10968–10969. [Google Scholar] [CrossRef]
- Taguchi, H.; Takeuchi, K.; Tsujimoto, S.; Matsuo, T.; Tanaka, H.; Yoshizawa, K.; Ozawa, F. Unsymmetrical PNP-pincer type phosphaalkene ligands protected by a fused-ring bulky Eind group: Synthesis and applications to Rh(I) and Ir(I) complexes. Organometallics 2016, 35, 1526–1533. [Google Scholar] [CrossRef]
- Takeuchi, K.; Tanaka, Y.; Tanigawa, I.; Ozawa, F. Cu(I) complex bearing a PNP-pincer-type phosphaalkene ligand with a bulky fused-ring Eind group: Properties and applications to FLP-type bond activation and catalytic CO2 reduction. Dalton Trans. 2020, 49, 3630–3637. [Google Scholar] [CrossRef]
- Yoshifuji, M. Low coordinate phosphorus compounds for 40 years in memory of the late Professor François Mathey, École Polytechnique. Phosphorus Sulfur Silicon Relat. Elem. 2021, 1–6. [Google Scholar] [CrossRef]
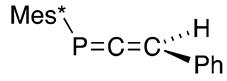
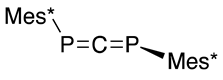
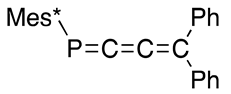
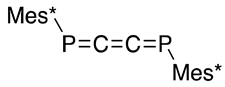
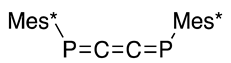
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).